同位素稀释液相色谱-串联质谱法测定小麦粉中黄曲霉毒素B1的不确定度评定
2020-09-02杨涛,袁辉
杨 涛,袁 辉
(新疆产品质量监督检验研究院,新疆 乌鲁木齐 830011)
黄曲霉毒素是主要由黄曲霉、寄生曲霉产生的次生代谢产物[1],在湿热地区食品和饲料中出现黄曲霉毒素的几率很高。它们存在于土壤、动植物、各种坚果中,特别是容易污染花生、玉米、小麦等粮油产品,是霉菌毒素中毒性最大、对人类健康危害极为突出的一类霉菌毒素。
黄曲霉毒素 B1是黄曲霉毒素的产物中对人体最具威胁的物质,具有致癌、致突变和致畸形作用。目前,检测黄曲霉毒素B1的方法主要有薄层色谱法[2], 高效液相色谱法和液相色谱-质谱联用法[3-4],以及酶联免疫吸附测定法[5]等。本研究按照2016年12月发布的GB 5009.22—2016《食品安全国家标准 食品中黄曲霉毒素B族和G族的测定》[6]中第一法:同位素稀释液相色谱-串联质谱法,对小麦粉中黄曲霉毒素B1进行测定,并依据 CNAS—GL 06《化学分析中不确定度的评估指南》和 JJF 1059.1—2012《测量不确定度评定与表示》[7-8]规定的方法建立了数学模型,分析了液相色谱-串联质谱法测定小麦粉中黄曲霉毒素B1过程中所引入的不确定度来源,并对各个不确定来源进行了分析、量化处理,得出小麦粉中黄曲霉毒素B1含量的扩展不确定度,对其最终测定结果进行不确定度评估,为小麦粉中黄曲霉毒素B1的测定结果的准确性提供一定的依据。
1 材料与方法
1.1 材料与试剂
黄曲霉毒素B1标准品,含量标示为(100.54±0.3)μg/ml,纯度为99%,Pribolab公司;同位素内标13C17-黄曲霉毒素B1标准品,含量标示为(0.501 ± 0.008) μg/ml,纯度为99.3%,Pribolab公司;甲醇,色谱纯,Fisher公司;乙酸铵,色谱纯,sigma公司;黄曲霉毒素B1免疫亲和柱,华安麦科;小麦粉为市场购买。
1.2 仪器与设备
AB 4000+高效液相色谱-质谱/质谱仪,美国AB公司; 色谱柱:岛津C18色谱柱,150 mm×2.1 mm,3 μm;Sartorius BSA22025电子天平,感量0.01 g,德国Sartorius公司;固相萃取装置,美国J2 Scientific公司;5418离心机,德国Eppendorf;超纯水机,美国millipore公司。
1.3 方法
1.3.1标准溶液的配制
移取黄曲霉毒素B1标准品1.00 ml于10 ml容量瓶,甲醇定容为标准储备液。移取储备液0.5 ml于50 ml容量瓶,定容为标准工作液。移取13C17-黄曲霉毒素B1标准品2 ml于10 ml容量瓶,作为同位素内标工作液。准确移取工作液10、100、300、500、1 000 μl至10 ml容量瓶中,加入200 μl同位素内标工作液,甲醇定容至刻度,得到内标质量浓度为2 ng/ml的标准使用液。
1.3.2样品前处理
准确称取试样5.00 g于离心管中,加入100 μl内标,震荡后静置30 min。加入(7+3)甲醇水20 ml,超声提取20 min。6 000 r/min离心10 min,取上清液4 ml,加25 ml水稀释,将待净化液注入免疫亲和柱中,调节压力以6 ml/min的流速过免疫亲和柱,直至2~3 ml空气通过柱体。准确加入1 ml色谱甲醇洗脱,流速为1~2 ml/min,收集全部流出液于样品瓶中,经0.22 μm滤膜过滤,供测试使用。
1.3.3仪器分析条件
流动相为5 mmol乙酸铵溶液∶甲醇,流速0.3 ml/min,柱温40℃,进样量:10 μl。质谱条件:电喷雾正离子扫描模式,多反应监测模式(MRM)见表1。

表1 多反应监测条件
1.3.4数学模型的建立与不确定度来源的分析
1.3.4.1不确定度的评估
不确定度的评估需要明确测量值的数学模型,进而分析其可能的不确定来源。本实验中黄曲霉毒素B1的数学计算模型如式(1)所示:
(1)
式中,X为试样中黄曲霉毒素B1的含量,μg/kg;m为试样质量,g;V1为 样品提取液总体积,20 ml;V2为免疫亲和柱上样体积,4 ml;V3为样品定容体积,1 ml;c为从标准曲线上测得试液中黄曲霉毒素B1的质量浓度,ng/ml;f为样品加标回收率,%。
1.3.4.2不确定度分量的主要来源及其分析
该方法前处理过程主要为手工操作,受人为因素影响较大。根据检测方法和实际操作情况,高效液相色谱法测定黄曲霉毒素B1不确定度主要有以下几个来源:一是标准溶液引入的不确定度,包括标准物质纯度引入的不确定度,标准溶液稀释配制过程引入的不确定度,最小二乘法拟合标准曲线产生的不确定度;二是样品前处理过程引入的不确定度,包括样品称量、提取溶剂的总体积、免疫亲和柱上样体积和最终定容体积以及提取效率和样品回收率引入的不确定度。
2 结果与分析
2.1 标准溶液引入的不确定度(c)[9-11]
黄曲霉毒素B1标准溶液引入的不确定度主要由标准物质自身纯度、标准工作液稀释配制过程以及标准曲线拟合等引入。
2.1.1标准物质自身纯度引入的不确定度(s)
根据标准证书可得知,黄曲霉毒素B1和同位素内标13C17-黄曲霉毒素B1的含量分别为100.54±0.3 μg/ml和0.501±0.008 μg/ml(k=2),按照均匀分布计算,由标准物质纯度引入的相对不确定度分别为:
urel(s1)=0.03÷(100.54×2)=0.001 49
urel(s2)=0.008÷(0.501×2)=0.007 98
因此,由标准物质纯度引入的不确定度为:
2.1.2标准溶液稀释过程中移液器和玻璃量器引入的不确定度(D)
2.1.2.1标准溶液稀释过程中体积引入的不确定度(V)
标准溶液配制的过程中使用了一系列的移液器和玻璃量器,依据JJG 196—2006《常用玻璃量器检定规程》和JJG 646—2006《移液器检定规程》[12-13]所明示,对本实验中所使用的移液器和玻璃量器均有相应的最大允许误差,按照矩形分布,标准溶液稀释过程中移液器和玻璃量器引入的相对不确定度见表2。
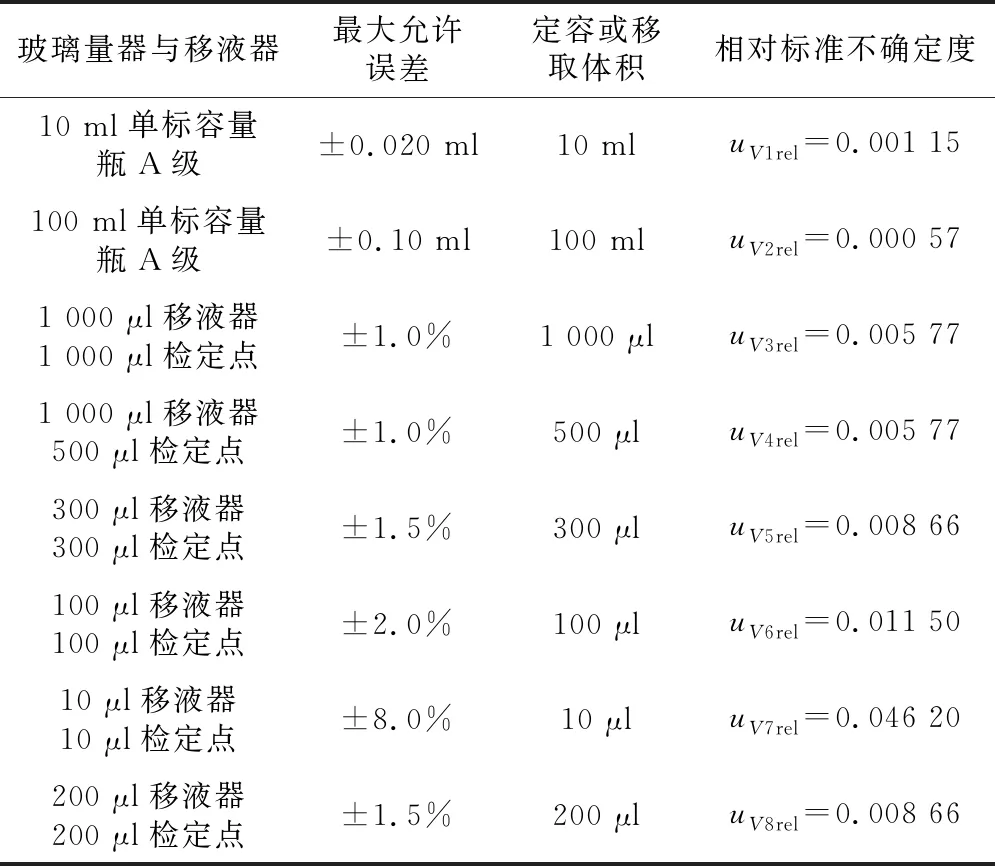
表2 标准溶液配制过程中玻璃量器和移液器引入的不确定度
在标准溶液稀释配制过程中,使用10 ml容量瓶7次,100 ml容量瓶1次, 1 000 μl移液器移取1 000 μl体积4次,1 000 μl移液器移取500 μl体积1次,300 μl移液器移取300 μl体积1次,100 μl移液器移取100 μl体积1次,10 μl移液器移取10 μl体积1次,200 μl移液器移取200 μl同位素内标体积5次。由表2中的数据合成标准溶液稀释过程中,移液器和玻璃量器引入的相对不确定度为:
=0.053
2.1.2.2标准溶液稀释过程中温度波动引入的不确定度(T)
移液器和玻璃量器的鉴定是在20℃下进行,配制标准溶液的温度要控制在(20±5)℃,因此,可以通过估算温度范围和体积膨胀系数,计算由温度波动引入的不确定度。由于玻璃的膨胀系数远远小于液体的膨胀系数,所以玻璃的膨胀系数可忽略不计。标准溶液是用甲醇稀释配制的,甲醇在20℃时的膨胀系数为1.19 ×10-3ml/℃,温度变化按照矩形分布,各移液器和玻璃量器因温度波动引入的不确定度见表3。

表3 标准溶液配制过程中温度引入的不确定度
根据表3中的数据,以及移液器和玻璃量器使用次数,合成标准溶液稀释过程中,温度变化引入的相对不确定度:
=0.015
因此,标准溶液稀释过程中移液器和玻璃量器引入的不确定度为:
=0.055
2.1.3最小二乘法拟合标准曲线引入的不确定度(q)
标液系列的质量浓度为0.1、1、3、5、10 ng/ml,内标的质量浓度均为2 ng/ml,在液相色谱-串联质谱仪上对五个校准标准溶液各测量3次,共计15次,得到各质量浓度标准物质所对应的峰面积比值。标准曲线数据如表4,用最小二乘法拟合各标准溶液质量浓度与峰面积比值的标准曲线。
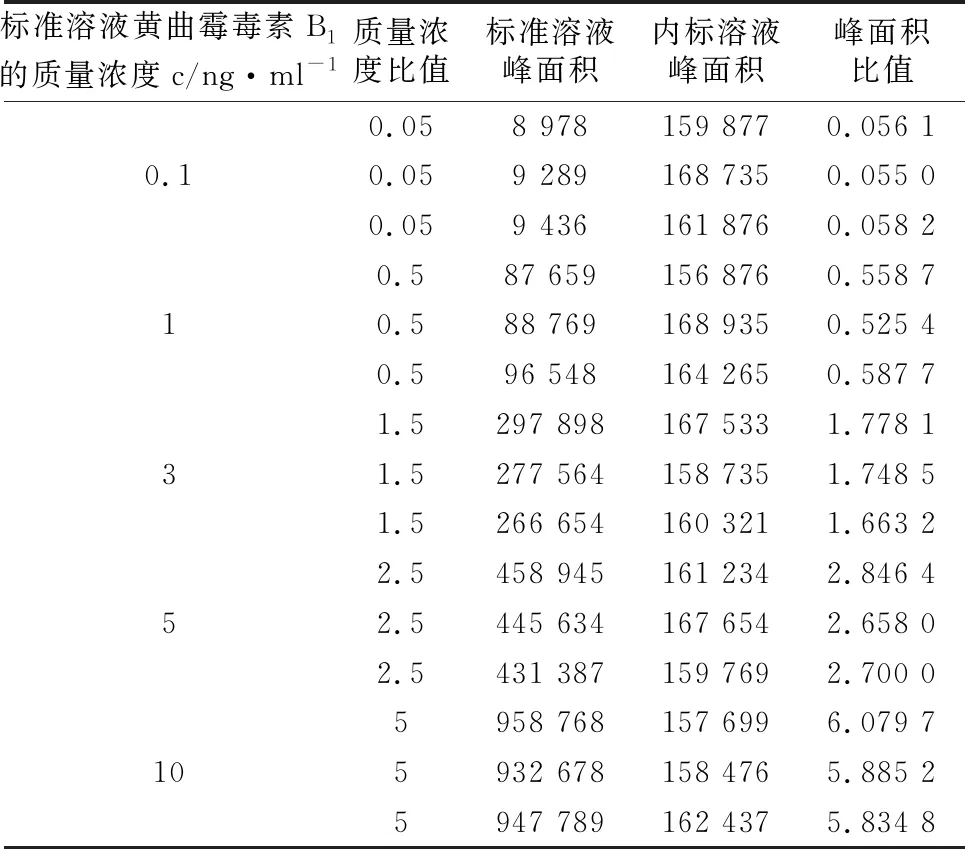
表4 内标法测定黄曲霉毒素B1标准曲线的数据
对被测样品溶液中的的黄曲霉毒素B1共测量2次,即p=2。根据峰面积,由标准曲线方程计算得到样品中黄曲霉毒素B1平均质量浓度为x=4.3 ng/ml。于是由最小二乘法拟合标准曲线引入的不确定度u(q)为
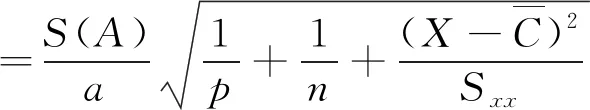
=0.176 6
其中:
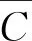
由标准曲线拟合的相对不确定度为:
urel(q)=u(q)/x=0.041
因此,样品在测定过程中,由标准溶液引入的不确定度为:
2.2 样品前处理过程引入的不确定度[10,14]
2.2.1样品称量过程引入的不确定度(m)
天平校准的允许最大误差为0.01 g,标准要求称样质量为5.00 g,按矩形分布原则,称样时由天平的最大允许误差导致的样品质量不确定度为
2.2.2添加内标溶液引入的不确定度(I)
采用100 μl移液器加入内标13C17-黄曲霉毒素B1标准溶液100 μl,由移液器引入的相对标准不确定度为:
urel(I)=uT6rel=0.011 5
2.2.3样品提取液总体积(V1)
样品提取液使用25 ml量筒量取(7+3)甲醇水20 ml,JJG196—2006《常用玻璃量器》[12]规定:20℃时25 ml量筒的容量允许误差为±0.25 ml,按矩形分布,则量筒量取提取液体积引来的不确定度分量为:
2.2.4免疫亲和柱上样体积(V2)
使用5 ml移液器移取4 ml提取液进行免疫亲和柱法,按照JJG 646—2006《移液器检定规程》[13]:20℃时5 ml移液器在5 ml检定点的最大允许误差为0.6%,按矩形分布,则免疫亲和柱上样体积引来的不确定度分量为:
2.2.5样品定容体积(V3)
样品最终定容使用1 ml单标线移液管移取色谱级甲醇进行定容,按照JJG 646—2006《移液器检定规程》[13]:20℃时1 ml移液器在1 ml检定点的最大允许误差为0.1%,按照矩形分布,则样品定容体积引来的不确定度分量为:
2.2.6提取效率和过小柱得到的回收率影响(f)
如1.3.2所述,测定小麦粉中的黄曲霉毒素B1的前处理需经过对样品的称量、提取、上柱净化、洗脱定容等步骤后经仪器分析测试。在这冗长的过程中必然会造成样品的测定值偏离真值。本实验对样品进行6次平行测试,根据6次结果进行计算,回收率结果见表5。
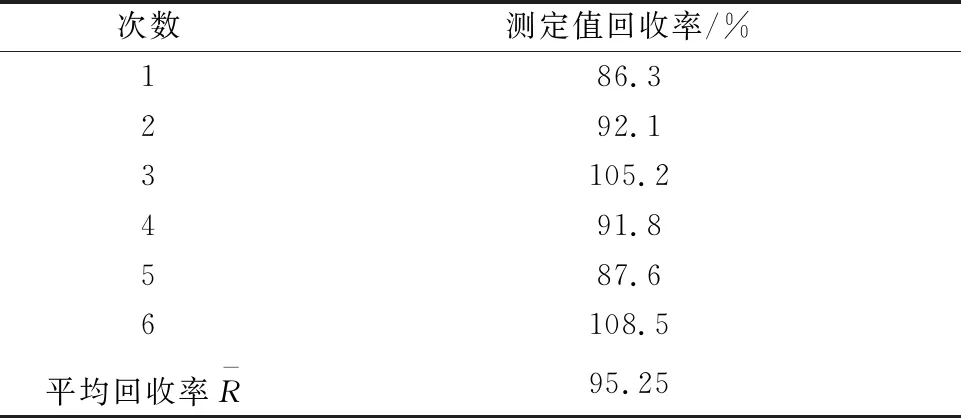
表5 回收率结果统计
根据表5的数据,回收率的相对标准偏差为4.0%,因此由样品回收率引入的相对不确定度为:
urel(f)=0.040
2.3 分析仪器引入的不确定度(E)
本实验所采用的超高效液相色谱-串联质谱仪的扩展不确定度为5%,k=2(由新疆自治区计量测试研究院出具的校准证书),则由液相色谱-串联质朴仪引入的相对不确定度为:
urel(E)=0.05÷2=0.025
2.4 测量不确定度的评定与报告
2.4.1合成标准不确定度
各相对不确定度分量见表6。
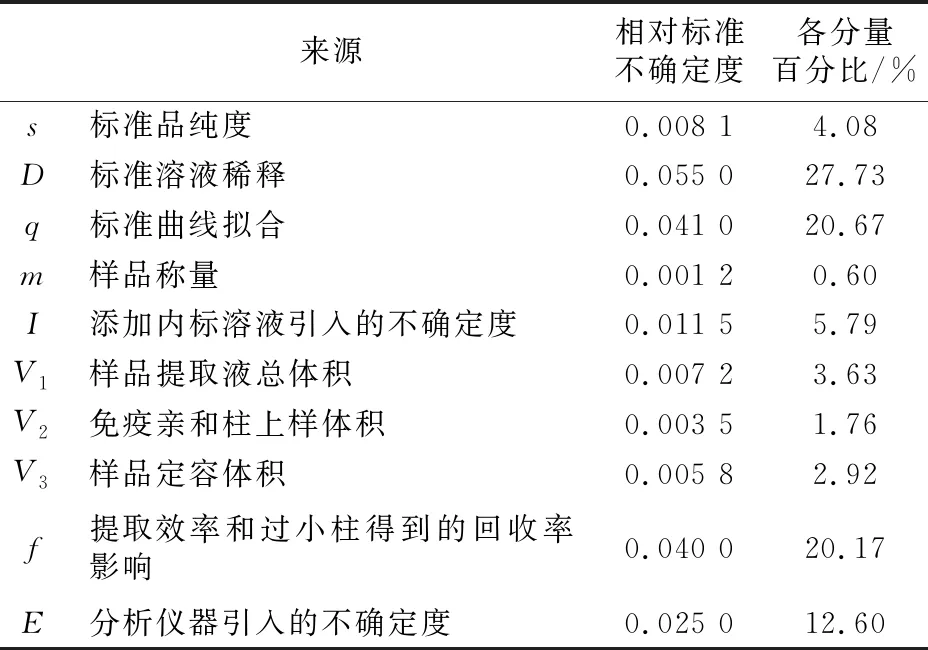
表6 各相对不确定度分量汇总表
由表6可以看出,小麦粉中黄曲霉毒素B1的相对标准不确定度分量贡献较大的分别是标准溶液的稀释配制、标准曲线拟合以及提取效率和过小柱得到的回收率,而样品称量、样品提取液体积等引入的相对标准不确定度相对较小。
由表6中的数据合成标准不确定度:
=0.084
2.4.2扩展不确定度
依据JJF1135—2005《化学分析测量不确定度评定》[15]规定,在95%置信水平下,取包含因子k为2,小麦粉中黄曲霉毒素B1的测量值为4.3 ng/ml,按照前处理过程,计算得到小麦粉中黄曲霉毒素B1的含量为4.30 μg/kg,则扩展不确定度
U= 4.30×0.084×2 =0.72 μg/kg
2.4.3测定结果
按照该方法测定小麦粉中黄曲霉毒素B1的结果为:X=(4.30±0.72)μg/kg,k=2。
3 结论
在报告测量的结果时,必须给出相应的不确定度,一方面便于使用它的人评定其可靠性,另一方面也增强了测量结果之间的可比性。
本研究根据实验过程建立数学模型,对小麦粉中黄曲霉毒素B1测定过程中的各个不确定度来源进行了量化分析。结果发现,影响黄曲霉毒素B1测定的不确定度的主要因素,主要来源于标准溶液稀释过程,其百分比高达27.73%;其次是最小二乘法标准曲线拟合以及提取效率和过小柱的回收率引入的不确定度,均在20%以上;而样品称量以及前处理过程中定容体积等引入的不确定度相对而言都比较小。
看似对检测结果影响不大的标准溶液稀释过程,因为有内标的加入,稀释的步骤越多,所产生的不确定度越大,从而增大了不确定度因素。因此,在小麦粉中黄曲霉毒素B1测定过程中,要着重加强不确定分量中百分比最大的三个方面的质量控制,从而降低测量不确定度,使测定结果的准确性得以提高。
