空气中氮气对危险废物氮元素测定结果的影响
2020-09-02鄢大彬任强肖伟骆岚古小林周晓润王茜杨思会刘威罗成琪
鄢大彬任强肖伟骆岚古小林周晓润王茜杨思会刘威罗成琪
( 1. 成都兴蓉环保科技股份有限公司,四川成都 610017; 2. 四川弗里曼环境检测有限公司,四川成都 610500)
1 引言
危险废物是指列入国家危险废物名录或者根据国家规定的危险废物鉴别标准和鉴别方法认定的具有危险特性的固体废物[1]。 危险废物必须进行利用和处置, 危险废物的利用是指从固体废物中提取物质作为原材料或者燃料的活动, 处置是指将固体废物焚烧和用其他改变固体废物的物理、化学、生物特性的方法,达到减少已产生的固体废物数量,缩小固体废物体积,减少或者消除其危险成分的活动,或者将固体废物最终置于符合环境保护规定要求的填埋场的活动。 企事业单位产生的危险废物需要交有资质的企业进行处置和利用。 危险废物处置技术包括焚烧处置技术、非焚烧处置技术、安全填埋处置等[2]。其中焚烧是重要的处置技术之一, 危险废物焚烧系统烟气排放要求严格, 其中氮氧化物是最容易超标的烟气指标之一,指标要求低于500 mg/m3[3]。 危险废物处置企业需要对进炉物料进行配伍, 限制物料氮元素含量以满足烟气控制标准。 这就要求企业必须对入场废物的氮元素含量进行检测, 得到各种废物氮元素含量后进行配伍焚烧。 目前,危险废物氮元素测定还没有相应的国家或行业标准, 行业内测定氮元素含量的方法主要是量热仪氧弹燃烧后吸收液吸收燃烧气体, 再采用离子色谱法测定硝酸盐和亚硝酸盐的方法测定氮含量。张爱平、王红卫等采用高温氧化燃烧和过氧化钠吸收做前处理, 采用离子色谱法同时测定石油焦样品中的硫、氮、氟、氯含量,氮元素本底值0.02%左右,加标量0.02%时,回收率可达80%以上[4]。 王思瑜、袁明华采用氧弹燃烧-离子色谱法测定龙葵中氯、氮、硫的含量也显示相似的检测结果[5],说明该方法在氮元素测定中是具备一定应用可能的。该方法在装样完成时,氧弹中会封闭一定空气,空气中氮气、氧气与后面充入的高压氧气混合在一起,氮气虽性质稳定,但在这种高压和瞬间燃烧过程会参与燃烧产生氮氧化物, 影响氮元素测试结果。
本文通过对比实验, 探讨了空气中氮气对氮元素测定过程影响的大小、控制及消除的方法,以期为危险废物处置企业氮元素测定提供参考。
2 实验部分
2.1 实验材料
成都市某危险废物处置企业入场废物样品包括固态( 活性炭、漆渣)、半固态( 精馏残渣、有机污泥)、液态( 废有机溶剂),每次取样量不少于1 kg,取样后混匀。该企业焚烧系统采用回转窑焚烧危险废物,固废、液废分别进料,烟气采用湿式、干式脱酸、非催化脱硝、布袋除尘进行烟气处理,设计焚烧量30 t/d。活性炭、漆渣样品为成都某汽车企业危废入场样品,精馏残渣及有机污泥为成都某制药企业危废入场样品,废有机溶剂为某制药企业入场样品。
2.2 仪器与试剂
ZDHW-HN7000 微机全自动量热仪( 鹤壁市华能电子科技有限公司)、 ICS-900 离子色谱仪( 赛默飞世尔科技)、M2002E 分析天平( 梅特勒-托利多,精度为0.000 1 g)、水平震荡器、分析纯氢氧化钠、纯度>99.99%氧气、优级纯硝酸钠、优级纯亚硝酸钠、在线生成( 0.025 mol/L KOH)淋洗液。
2.3 样品制备
样品的采集和制样按照《 工业固体废物采样制样技术规范( HJ/T 20—1998)》操作。 试样制备时,固体废物样品应取不少于500 g 于洁净研钵中捣碎、混合均匀。若为尺寸较大样品,用剪刀或破碎机破碎后混合均匀。膏状样品取样不少于500 g 于1 L 烧杯中用玻棒搅拌不少于3 min,充分混合均匀。 液态样品取样时应根据固体废物在容器中的情况而定,若无分层情况,取样前应充分震荡均匀( 因焚烧类液体废物挥发性通常较强,不宜用搅拌方式混匀);若有分层情况,应静置1 h 以上,待分层明显后,分层取样,分别测试。
2.4 实验方法
样品称量时, 固体和膏状样品直接用勺子或镊子将样品移取到与氧弹配套坩埚中, 样品质量控制在0.05~0.1 g 之间,精确至0.1 mg。 液态样品称量前应先称量坩埚及胶囊( 不含氮元素)质量,采用2.5 mL 带钢头进样针抽取样品后迅速注入胶囊长度1/3 处,样品质量控制在0.05~0.1 g 之间,记录样品质量。加入助燃剂固态苯甲酸一粒,记录助燃剂质量,同时,保证助燃剂与样品或胶囊在坩埚中接触良好。
装样及燃烧测定时应迅速将装有样品和苯甲酸的坩埚装入氧弹中( 苯甲酸单独测量时,只加苯甲酸即可), 氧弹底部应提前装入0.2%的氢氧化钠吸收液10 mL,双氧水0.5 mL,固定好点火丝,使点火丝与苯甲酸接触良好,拧紧氧弹盖,在氧弹加气装置充氧至3.0±0.1 MP, 将氧弹排气处与尾气吸收装置连接,吸收液采用去离子水,排气至吸收装置无气泡时停止排气。 重复以上充氧排气过程, 在第3 次充氧后,将氧弹放入量热仪中测定热值。若非置换方式测定时,氧弹中充氧后直接放入量热仪中测试即可,不用置换过程( 连续2 次充装高压氧气并排气后,可将原氧弹中的空气中的氮气置换排出99%以上)。
吸收液收集时,热值测试完成5 min 后取出氧弹,放置在水平震荡器上,常温,100 r/min,震荡10 min,震荡完成。将氧弹排气处与尾气吸收装置连接。尾气吸收装置应至少为二级吸收,吸收液为0.2%氢氧化钠溶液。 缓慢释放压力,排气速率均匀,在吸收装置中仅观察到小气泡为宜。 在排气至吸收装置处观察不到气泡后,继续按压排气10 s 停止。将氧弹在充气装置上充氧至3.0±0.1 MP,持续5 s。 连接氧弹排气处与尾气吸收装置,重复以上尾气排气操作,尾气吸收完成。 打开氧弹,将氧弹内吸收液转移至容量瓶,采用20 mL 吸收液仔细冲洗氧弹的内部、端子、氧弹盖的内表面和坩埚等,再用去离子水冲洗以上部位,冲洗次数不少于3 遍,将收集的氧弹吸收液、尾气吸收液、 全部冲洗液经孔径0.45 μm 微孔滤膜抽滤后定容至100 mL,待离子色谱测定。
离子色谱检测时按照说明书操作仪器, 仪器工作条件如下: 淋洗液采用0.025 mol/L KOH, 流速1.00 mL/min,抑制器电流45 mA,进样量10 μL。
校 准 曲 线 的 绘 制 配 制 浓 度 为1.00,2.00,5.00,10.00,15.00,25.00,50.00 mg/L 的硝酸根和亚硝酸根标准系列, 将上述标准系列从低浓度至高浓度依次进样,进样体积为10 μL,得到不同硝酸根和亚硝酸根色谱图。
以硝酸根和亚硝酸根浓度( mg/L)为横坐标、峰面积( 或峰高)为纵坐标,绘制校准曲线。按绘制校准曲线相同的色谱条件和步骤,进行试样的测定,记录样品的峰面积( 或峰高)。
固体废物氮含量按照下列公式计算:
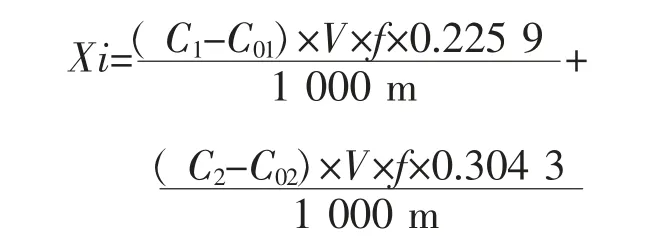
式中,Xi 为固体废物样品中氮含量,g/kg;C1为检测到的硝酸根含量浓度,mg/L;C2为检测到的亚硝酸根含量浓度,mg/L;C01为空白实验中硝酸根含量浓度,mg/L;C02为空白实验中亚硝酸根含量浓度,mg/L;V为吸收液的定容体积,mL;f 为稀释倍数;m 为称量固体废物样品质量,g;0.225 9 为硝酸根中氮含量系数;0.304 3 为亚硝酸根中氮含量系数。
依据以上方法, 分别在氧气置换与非置换情况下,测定苯甲酸、废活性炭+苯甲酸各5 次,其他危险废物样品各2 次,测试结果见表1。

表1 氧气置换与非置换检测结果对比
3 结果与讨论
3.1 空气中氮气对氮元素检测结果具有明显影响
表1 中苯甲酸单独燃烧氮元素检测结果显示,非高压氧气置换情况下, 最高39.78 mg/kg, 最低38.83 mg/kg,平均值39.30 mg/kg;高压氧气置换排除空气中氮气影响情况下, 氮元素检测结果最高0.055 mg/kg,最低0.046 mg/kg,平均值0.051 mg/kg。从检测结果可以看出, 空气中氮气对该种检测方法测定氮元素结果有明显的正影响, 由于氮气参与反应,检测结果明显偏高。实践中可以采用多次测定扣除背景的方式予以校正。
3.2 参与反应的氮气随着燃烧物质的增加影响增大
从表1 中废活性炭与苯甲酸对比检测结果看( 样品编号2-1 至2-5), 随称量的危险废物的量的增加,氮元素检测结果有明显的增加趋势。
参与燃烧的物质的量越多, 氧弹中参与反应的氮气的量越大, 这可能是由于参与燃烧的物质量越多,产生的热量越大,影响分布于氧弹内坩埚周围的氮气范围越广有关。
3.3 危险废物氮元素含量较低时,氧气多次置换方式可以有效减少空气中氮气的影响
从表1 中苯甲酸单独燃烧测量和废活性炭+苯甲酸混合燃烧检测结果看, 高压氧气置换情况下都明显低于非置换状态氮元素含量, 说明采用氧气置换方式是可以有效减少空气中氮气影响因素的。 精馏残渣+苯甲酸、有机污泥+苯甲酸、废有机溶剂+苯甲酸混合燃烧测定时,由于氮元素含量较高,氧气置换与非置换情况对比检测结果差异不明显。
