遗传性脑小血管病诊疗的研究进展
2020-08-29郭蕾江凌玲陈玮琪王伊龙
郭蕾 江凌玲 陈玮琪 王伊龙
脑小血管病(CSVD)是由各种原因引起脑内小动脉、微动脉、毛细血管、微静脉和小静脉受累所导致的临床、影像和病理综合征[1],占卒中病因的20%、痴呆病因的45%[2-3],是一种与年龄相关的常见疾病。 随着分子遗传学的发展,多种引起CSVD 的致病基因被发现,CSVD 继而被粗略地分为散发性和遗传性CSVD,其中遗传性CSVD 约占5%[4]。 与散发性CSVD 相比,遗传性CSVD 在基因特点、临床分类、影像学和病理学表现方面不完全相同,因此在诊疗方面也有区别。 然而,除Fabry 病(FD)外,目前尚无针对遗传性CSVD 的有效临床治疗手段。 本文结合国内外指南及相关文献,对目前研究较多的遗传性CSVD 进行综述,概述其近年诊疗方面的研究进展。
一、遗传性CSVD 分类
根据累及小血管病因及病理改变,CSVD 被分为6 大类,其中与遗传相关的两类为遗传性脑淀粉样血管病(CAA)和其他除外CAA 的遗传性CSVD[如伴皮质下梗死和白质脑病的常染色体显性遗传性脑动脉病(CADASIL)、伴皮质下梗死和白质脑病的常染色体隐性遗传性脑动脉病(CARASIL)、伴卒中和白质脑病的组织蛋白酶A 相关性动脉病(CARASAL)、FD、视网膜血管病变伴白质脑病和多系统损害(RVCL-S)及COL4A1/2 相关 CSVD 等[1]。 不同遗传性 CSVD 在病理改变、累及的小血管结构及病理生理机制方面表现出不同的特征(表1)。 但是,数项经典遗传性CSVD的研究表明,神经血管单元完整性破坏(包括细胞外基质紊乱)是致病的主要途径[5]。
二、各类遗传性CSVD 的诊疗进展
1.CADASIL
CADASIL 是NOTCH3 基因(Chr19p13.12)突变导致的常染色体显性遗传性脑小动脉病,是最常见的一种遗传性CSVD。NOTCH3 基因编码跨膜受体蛋白主要在血管平滑肌细胞和周细胞上表达[6]。 CADASIL患者可观察到受累小血管平滑肌细胞萎缩和变性[7],临床上表现为先兆偏头痛、复发性卒中、精神症状及认知功能下降等。 60% ~85%的患者出现短暂性脑缺血发作和脑梗死;60%出现以执行功能受损为主的认知功能下降,并随年龄增长和卒中的反复发作而恶化[8],最终进展为残疾和痴呆。 CADASIL 患者的典型影像学表现见图1。
CADASIL 的致病突变集中在NOTCH3 蛋白类表皮生长因子重复序列(EGFR)区域,主要引起半胱氨酸残基数量不均[9]。 突变基因检测具有近乎100%的特异性和敏感性,为诊断金标准。 电镜观察血管平滑肌细胞周围颗粒状嗜锇物质(GOM)沉积及NOTCH3蛋白免疫组化染色亦具有较高特异性和敏感性[10-11]。最新诊疗共识提出,NOTCH3 突变检测为CADASIL 的确诊依据;当基因检测显示临床意义未明突变时,皮肤活检可作为诊断手段;对于不明原因的皮质下梗死、广泛对称性白质病变和基底节区多发性腔隙性梗死,且伴先兆偏头痛、复发性缺血性卒中或痴呆家族史者,应怀疑CADASIL[12-13]。
CADASIL 尚无特异性治疗方法,目前主要给予对症治疗。 偏头痛治疗可遵循常规临床指南。 利培酮、丙戊酸钠等对精神症状可能有治疗作用[14]。 对于伴有认知障碍的患者,多奈哌齐可能对其执行功能有改善作用[15]。 吸烟、高血压等传统血管危险因素与CADASIL 卒中和偏头痛风险增加有关[16-17],可建议患者戒烟及控制血压。 对于小血管急性缺血性卒中患者不应给予溶栓治疗,除非并发大动脉梗阻[12]。 短期阿托伐他汀治疗未能改善CADASIL 脑血流[18],因此对于胆固醇正常者不推荐应用他汀类药物[12]。 随着机制研究的深入,有学者提出通过外显子跳跃去除突变的EGFR 区域,以消除突变NOTCH3 的累积及一系列负性级联反应[19]。 此外,研究发现干细胞因子和粒细胞集落刺激因子可改善 CADASIL 小鼠的认知功能[20],干细胞治疗可能成为CADASIL 最有前景的治疗手段之一。
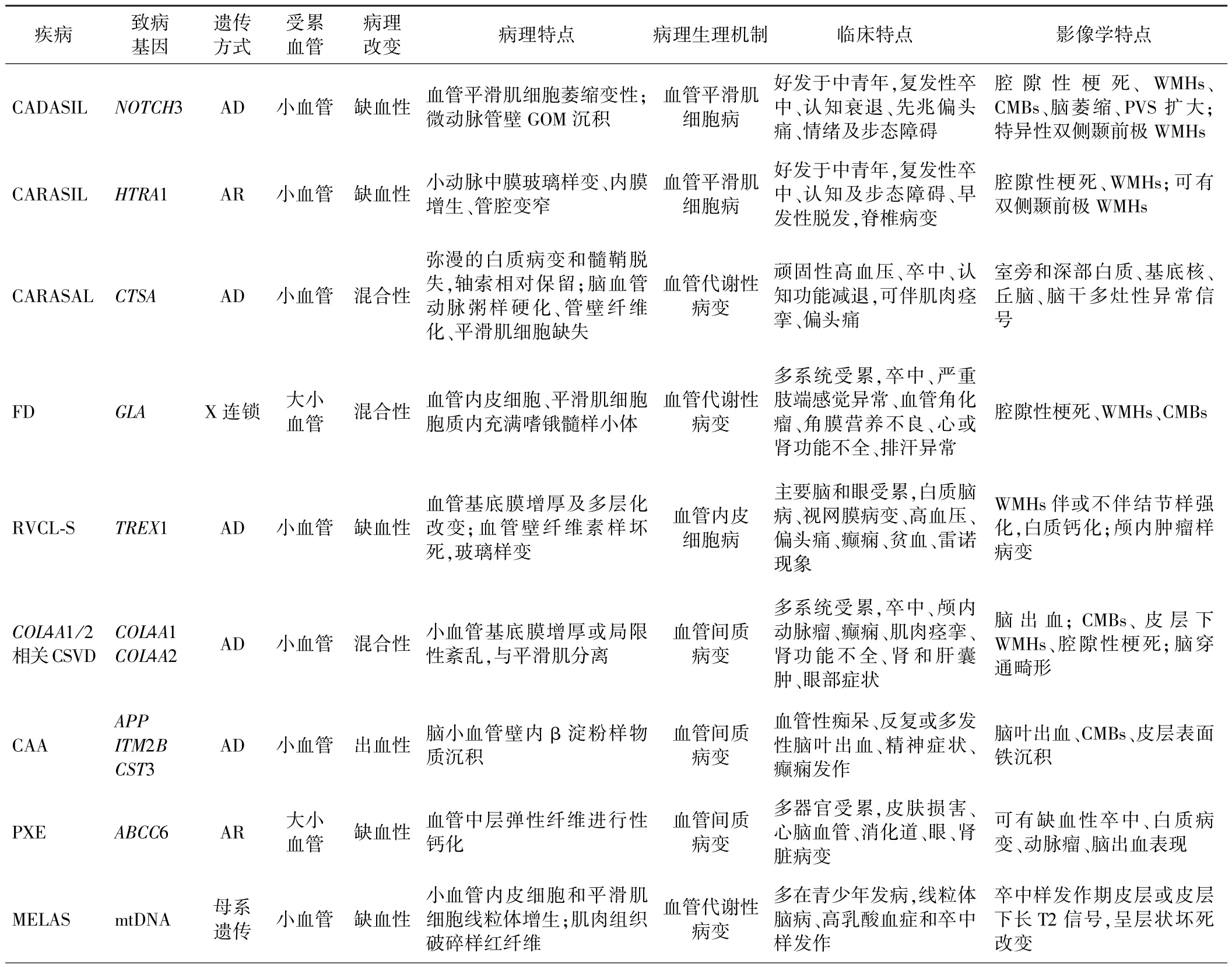
表1 主要遗传性CSVD 的特征
2.CARASIL
CARASIL 是由丝氨酸蛋白酶(HTRA1) 基因(Chr10q26.3)纯合突变所致。HTRA1 纯合突变可引起转化生长因子(TGF)-β 信号通路异常,导致常染色体隐性遗传性CSVD[21];而HTRA1 杂合突变则导致常染色体显性CSVD[22]。 尸检发现,CARASIL 患者脑和脊髓小血管出现内膜增生、玻璃样变性、管腔变窄等小动脉硬化表现[23]。 患者可表现为早发性脱发、严重腰背痛、进行性加重的痴呆和步态障碍,部分患者亦出现复发性腔隙性卒中、情绪异常、假性球麻痹、脊椎病等,平均病程 20 ~30 年[24]。 CARASIL 患者的典型影像学表现见图2。
电镜下CARASIL 患者脑标本中病变血管几乎未观察到GOM 存在[23]。 其他器官的小血管病变与脑血管相比更为轻微且无特征性改变[25],因此皮肤或肌肉活检无法诊断。 CARASIL 诊断主要依靠基因检测,其检出率 >95%[21]。 CARASIL 诊断要点与 CADASIL 相似:HTRA1 纯合突变为确诊依据;对于不明原因反复发作的腔隙性卒中伴严重WMHs 的病例,尤其与早发性脱发、腰痛或脊椎病相关者,应怀疑CARASIL[12-13]。
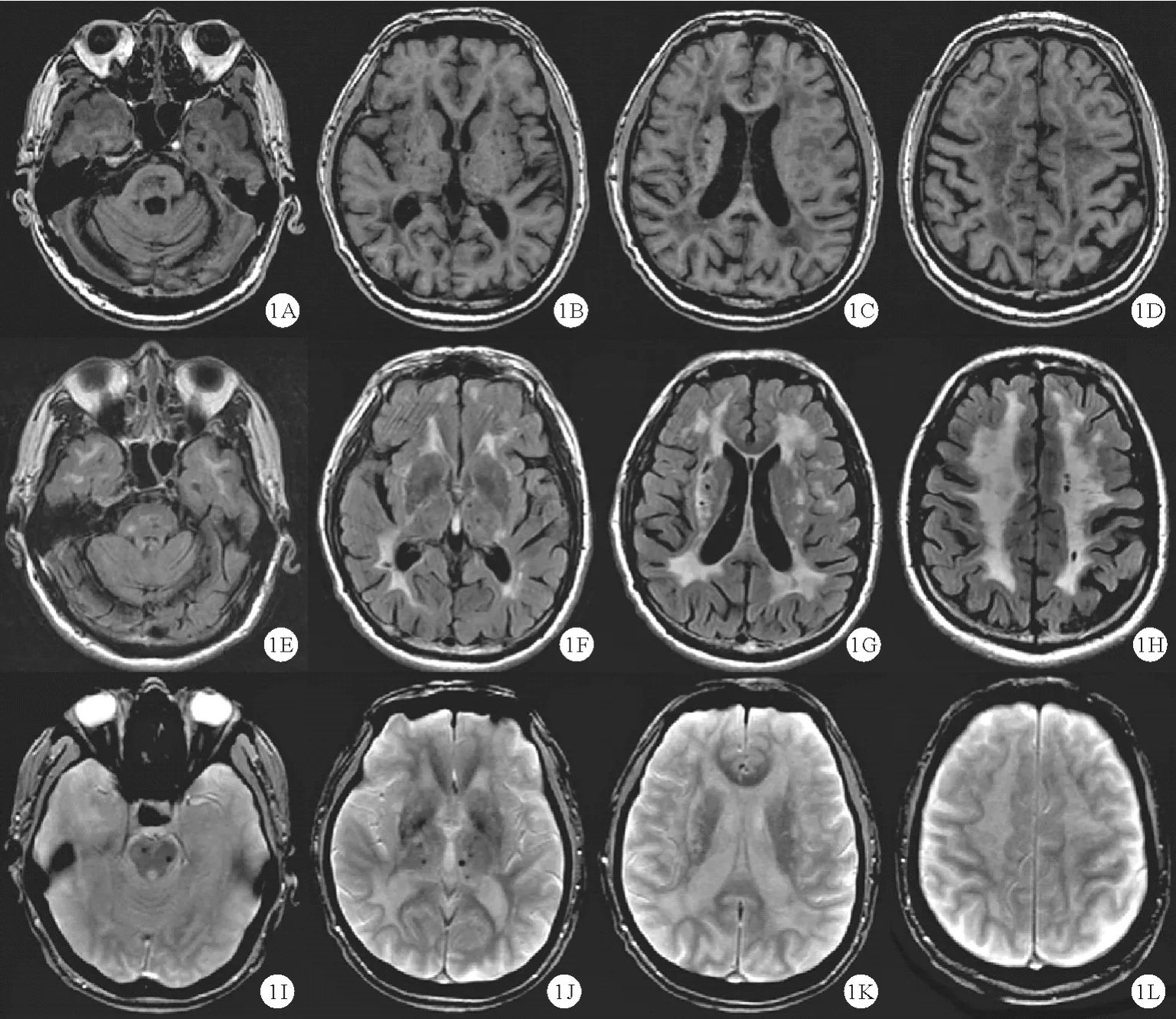
图1 CADASIL 患者影像学表现[8]:A ~ D:MRI 显示 T1WI 脑干、基底节、豆状核腔隙性梗死;E ~ H:FLAIR 像深部小梗死灶及颞前极融合的WMHs;I ~L:T2∗WI 脑干及丘脑微出血
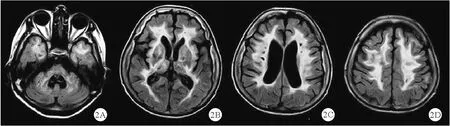
图2 CARASIL 患者影像学表现[24]:FLAIR 像广泛白质病变,包括颞叶前部受累,伴室周和丘脑多个腔隙灶及内囊、外囊高信号
CARASIL 目前无特效治疗方法。 对于脑微出血患者,临床常给予抗血小板聚集和降压治疗,但无充足证据支持其疗效[12]。 步态障碍可进行物理康复训练,有助于改善协调困难。 痉挛状态可考虑应用巴氯芬、替托尼定等药物[26]。 情绪障碍和脊椎病的治疗遵循专科标准治疗。 血管危险因素会加重CADASIL 病情,亦可能对其他单基因遗传性CSVD 有类似作用,因此建议患者戒烟及控制血压。 基于对发病机制的研究,增强HTRA1 或降低TGF-β 活性等针对性治疗策略被提出,用于其他疾病如马凡综合征的TGF-β 信号通路抑制剂也被考虑用于CARASIL 治疗[24]。
3.CARASAL
CARASAL 是CTSA基因(Chr20q13.12)突变引起的常染色体显性遗传病。CTSA基因编码组织蛋白酶A,突变可影响其活性从而减少内皮素-1 裂解,导致血管收缩及脑组织缺氧,表现出白质病变和顽固性高血压[27]。 CARASAL 的常见发病年龄为 30 ~ 40 岁,患者可表现为顽固性高血压、卒中、认知功能减退三联征,亦可出现发作性头痛、步态异常、肌肉痉挛等。 MRI 检查结果表现为类似散发性CSVD 的特征,以室旁和深部白质、基底核、丘脑、脑干等处信号改变为主,微出血、腔隙并不常见[27-28]。
CARASAL 应综合临床表现、影像学检查和CTSA基因检测进行诊断。 对于有卒中家族史、认知障碍、顽固性高血压表现及不明原因的广泛幕上或幕下白质和灰质高信号的CSVD 患者,应怀疑CARASAL。 目前,CARASAL 尚无特效治疗方法,可对患者出现的认知障碍、肌肉痉挛、偏头痛等予以对症处理,对于顽固性高血压进行监测及规范治疗[29]。
4.FD
FD 是α 半乳糖苷酶A(GLA)基因(ChrXq22.1)突变导致的X 连锁代谢疾病,GLA突变引起鞘脂类球藻神经酰胺(Gb3)及其去乙酰化衍生物(lyso-Gb3)在血管内皮细胞和平滑肌细胞、肾脏、心脏、背根神经节积累[30]。 与女性杂合突变患者相比,男性表型更重且发病更早,可在儿童期起病。 经典型FD 可出现严重肢端感觉异常、血管角化瘤、角膜营养不良、肾功能不全、左心室肥大、卒中等[30],卒中常在20 ~50 岁发病,86.8%为缺血性卒中[31]。 FD 患者影像学表现的典型影像学表现见图3。
GLA突变检测是诊断FD 的金标准,且有助于疾病表型确立及家系分析[32]。 男性患者亦可通过检测血浆、白细胞中α-GLA 活性诊断[33],但30%的女性患者酶活性可正常[34],因此女性不能据此诊断FD。 电镜下足细胞、肾小管上皮细胞、心肌细胞、血管内皮细胞和平滑肌细胞等胞质内充满嗜锇髓样小体为FD 特征性病理表现[33],有助于 FD 诊断。 此外,lyso-Gb3、hsTNT 等生物标志物可能有助于FD 的诊断和分期[35]。
与其他遗传性CSVD 不同,酶替代疗法(ERT)通过体外合成α-GLA 替代体内缺陷酶可特异性治疗FD。 多项临床研究结果显示,阿加糖酶可缓解FD 症状,改善受累器官功能及减少并发症[36-37],且尽早开始ERT 获益更大[36,38]。 酶增强治疗也是特效治疗方法,分子伴侣如米加司他可与突变的GLA结合,稳定其正常折叠结构,增强FD 患者的酶活性,改善肾脏等器官功能[39]。 最新诊疗共识提出,FD 不是系统性溶栓的禁忌证;血栓切除术可用于大脑动脉近端闭塞者;FD 患者首次发生脑血管事件后应接受抗栓治疗[32]。对于各器官损害应给予对症治疗,如应用卡马西平、苯妥英等缓解肢体疼痛[40];应用血管紧张素转化酶抑制剂(ACEI)或血管紧张素Ⅱ受体拮抗剂(ARB)减轻蛋白尿,严重者进行肾移植[32]。 此外,第二代ERT、底物还原治疗、基于mRNA 和基因等新的治疗方法也在研究中[41]。
5.RVCL-S
RVCL-S 是多器官系统受累的常染色体显性遗传病,由TREX1 基因(Chr3p21.3-21.2)杂合突变引起。RVCL-S 的主要特征为视网膜和脑微血管病变,常在30 ~50 岁发病,可表现出视力下降和视野缺损、偏瘫、偏头痛、认知障碍、精神障碍、癫痫、肝肾功能异常、高血压、贫血、雷诺现象等。 影像学呈现颅内肿瘤样病变,WMHs 伴或者不伴结节样强化、白质钙化等[42]。RVCL-S 患者的典型影像学表现见图4。
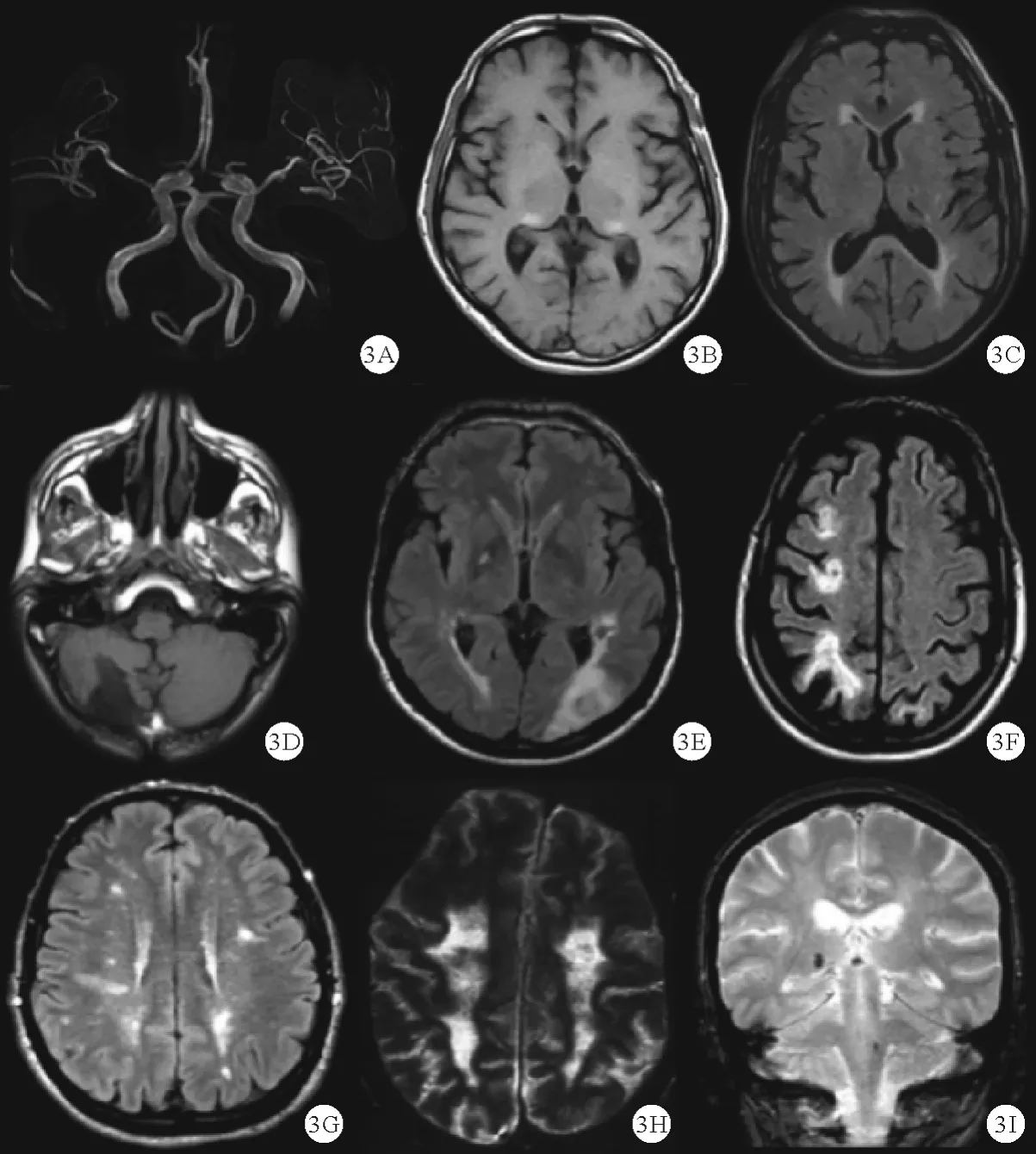
图3 FD 患者影像学表现[12]:A ~C:基底动脉扩张延长,双侧丘脑枕核T1 高信号,腔隙性卒中;D ~F:后循环缺血后脑软化,急性大脑中动脉脑梗死,分水岭脑梗死;G ~I:局灶性和融合性白质改变,弥漫性白质改变和萎缩,微出血
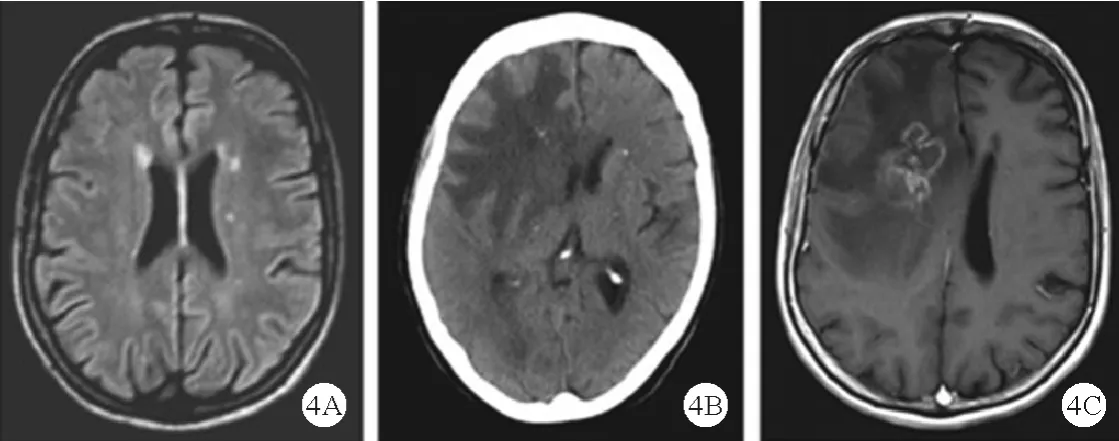
图4 RVCL-S 患者影像学表现[42]:A:深部及室周WMHs;B、C:右额叶边缘强化病灶,伴周围水肿、占位效应及钙化
RVCL-S 可通过TREX1 突变检测诊断,致病突变检出率>99%[43]。 对于有视觉障碍、神经功能损伤和相应影像表现且有家族史者应怀疑RCVL-S。 此外,血管性血友病因子(vWF)抗原、vWF 前肽和血管紧张素Ⅱ(AngⅡ)可作为疾病早期生物标志物[44]。
RVCL-S 尚无特异性治疗方法。 视网膜血管病变可进行激光治疗;黄斑水肿可应用贝伐珠单抗治疗;对于高血压、偏头痛、癫痫、贫血、消化道出血等给予相应的专科治疗[43]。 糖皮质激素可能减轻病灶周围的炎症及水肿[45],但目前尚无充足证据支持免疫抑制治疗的疗效。
6.COL4A1/2 相关CSVD
COL4A1/2 基因位于Chr13q34,编码Ⅳ型胶原α-1和2 链,参与构成包括脑小血管在内的血管基底膜。COL4A1/2 基因突变可导致胶原链沉积、基底膜缺陷和内质网应激[46]。 患者可表现出卒中、脑微出血、白质脑病、颅内动脉瘤、癫痫、肌肉痉挛、肾脏和眼部异常等广泛多变的表型[47-48]。 出血性卒中约是缺血性卒中的2 倍,脑出血在婴儿、儿童和成人中均可出现,且与活动、创伤和抗凝治疗有关[12,49]。
COL4A1/2 相关CSVD 的诊断应基于临床表型、家族史及遗传学检测。 当患者出现不明原因的深部脑出血、WMHs、脑穿通畸形,伴视网膜血管迂曲扩张、早期白内障、眼前节异常、婴儿偏瘫、颅内动脉瘤、肌肉痉挛、血尿、肾功能不全、肾或肝囊肿家族史,或出现早发性脑桥梗死时,应怀疑由COL4A1/2 突变引起[12]。
对于COL4A1/2 相关CSVD 的管理,最新诊疗共识不推荐静脉溶栓、抗血小板聚集或抗凝治疗;应避免时间长、强度高或有头部外伤风险的体育运动;COL4A1/2 突变的胎儿分娩时应考虑剖腹产[12]。 除促进突变COL4A降解外,减少突变蛋白胞内累积也可成为COL4A相关 CSVD 的治疗靶点。 研究发现4-苯基丁酸钠(4PBA)可减少胞内突变COL4A累积,缓解内质网应激[46],降低脑出血的发生风险[50]。
三、总结
遗传因素是CSVD 少见的病因之一,遗传性CSVD临床上常表现为多系统疾病,其症状、体征、影像学表现及实验室检查等与散发性CSVD 有所区别。 临床医生应加强对遗传性CSVD 的认识,关注发病年龄早、有家族史、典型临床和影像学表现的脑小血管病患者,对于疑诊患者进行分子遗传学或其他检测以明确或排除遗传性CVSD 的诊断,并给予相应治疗。
