磷掺杂碳催化剂的制备及其催化环己烷无氧脱氢性能
2020-08-26陈思远邢菲菲
周 丹,刘 博,陈思远,姜 标 ,邢菲菲,赵 虹*
(1.上海大学理学院,上海 200444; 2.中国科学院上海高等研究院,上海 201210)

氢能作为一种清洁、高效、可持续的新能源具有广泛的应用前景,目前安全高效的储氢环节是氢能应用的技术瓶颈之一。环己烷-苯是理想的有机液体储氢体系,质量储氢密度达7.19%,体积储氢密度为56 kg·m-3,且可以安全地实现大规模储存和远距离运输[8]。目前,苯加氢制环己烷的技术已相当成熟,开发更加高效的环己烷脱氢技术是解决环己烷-苯贮氢技术工业应用的关键。环烷烃脱氢是一个强吸热过程,通常需要在催化剂的作用下进行。常用的Pt、Pd等贵金属催化剂脱氢性能优异,但经济成本高而且易发生过度脱氢使催化剂结焦失活。而Ni基催化剂容易使C-C键断裂导致苯选择性较低[9]。开发高转化率、高选择性和高稳定性的环己烷脱氢催化剂将有利于推动氢能的规模化应用。
本文采用水热法制备磷掺杂的淀粉微孔碳催化剂(P@C),并应用于催化环己烷无氧脱氢制苯和氢气,系统考察磷酸用量对催化剂结构和催化剂脱氢性能的影响。
1 实验部分
1.1 催化剂制备
将15 g水溶性淀粉加入到80 mL去离子水中,搅拌均匀后置于带有聚四氟乙烯内衬的水热反应釜中,在180 ℃下水热处理8 h。取出水热反应釜,自然冷却至室温后打开反应釜,将水热处理后的物料过滤后,收集到的黑色固体置于80 ℃烘箱中干燥12 h后得到淀粉碳前驱体。将6 g前驱体和一定量的质量分数为85%的磷酸溶液分别加入到35 mL去离子水中,并在80 ℃下搅拌加热12 h。加热结束后将物料直接蒸干并在80 ℃烘箱中干燥12 h。将干燥后的样品置于管式电热炉中,在氮气气氛下以3 ℃·min-1的速率升温到800 ℃并保温5 h。获得磷掺杂淀粉碳催化剂样品,标记为P@C-X,其中X为磷酸添加量(P@C-2.5、P@C-5、P@C-10),不添加磷酸的前驱体直接碳化样品作为空白对比样记为Raw C。
1.2 催化剂表征
采用美国麦克仪器公司ASAP2020自动物理吸附仪对样品比表面积、孔径和孔分布等进行测定。测试前对催化剂样品脱气预处理,预处理温度90 ℃,脱气时间60 min,随后以10 ℃·min-1升温至350 ℃,脱气3 h后降温,然后在-196 ℃下吸附氮气。通过BET法计算碳材料比表面积,DFT法计算孔径分布,t-plot法计算孔体积。
X射线衍射测试采用日本理学公司Rigaku MiniFlex-600型多晶粉末衍射仪,Cu靶Kα射线辐射源,工作电压40 kV,工作电流40 mA,连续扫描速率0.5°·min-1,扫描范围5°~70°,步长0.02 °。
采用法国HORIBA公司Lab RAM-HR Evolution型拉曼光谱仪分析材料的缺陷程度,激光波长532 nm,光谱分辨率≤1 cm-1,扫描次数60次。
采用美国Thermo-VG Scientific公司ESCALAB250型X-射线光电子能谱仪对样品的组成、含量及化学状态进行分析。测试条件:Al Kα 射线,能量为1 486.6 eV,工作电压12 kV,工作电流20 mA。
氨气程序升温吸脱附分析采用美国麦克仪器公司ChemiSorb 2750型程序升温化学吸附仪进行测定。测试程序为在30 mL·min-1的氦气氛氛下,从室温以10 ℃·min-1的升温速率升至 650 ℃并恒温0.5 h,然后迅速降温至80 ℃,吸附氨气至饱和,氦气吹扫30 min,基线平稳后10 ℃·min-1升温至650 ℃。
1.3 催化剂性能评价
环己烷非氧催化脱氢制苯和氢气的反应在三段控温的固定床反应器中进行。石英反应管总长100 cm,内径10 mm,催化剂装填量为1.0 g。环己烷由神舟微科ZB-1L10A双柱塞微量泵泵入180 ℃气化室气化后,由载气N2带入催化剂床层进行反应。环己烷进料量为1 mL·h-1,N2流速为40 mL·min-1。采用岛津GC-2014C气相色谱仪在线分析反应器出口各有机物质组成,色谱柱为Agilent GS-Gaspro 30 m×0.32 mm,柱温60 ℃,进样器温度260 ℃,载气为高纯N2。因为反应产物相对单一,且少量副产物在色谱条件下均能检出,所以本实验中环己烷转化率及产物选择性采用面积归一法进行定量分析计算。H2采用Aglient 7890B气相色谱仪进行检测分析,色谱工作条件为TCD检测器设定温度为50 ℃,色谱柱型号为Porapak Q,载气为高纯Ar。
2 结果与讨论
2.1 磷酸用量对催化剂孔结构的影响
图1为Raw C、P@C-2.5、P@C-5、P@C-10碳材料催化剂样品的氮气吸附-脱附等温曲线和孔径分布图,其元素含量、比表面积、孔容和孔径数据见表1。由图1可知,氮气的吸附量在相对压力<0.1的低分压区急剧增大,并且在中分压和高分压区段吸附曲线和脱附曲线几乎完全重合,并没有观察到明显的滞后环,表明碳材料样品主要是以微孔为主的多孔结构。如表1所示,未加入磷酸改性的Raw C样品,比表面积为696.1 m2·g-1。经磷掺杂后,P@C-2.5、P@C-5和P@C-10样品的比表面积分别为1 194.9 m2·g-1、1 609.8 m2·g-1和1 963.2 m2·g-1,说明磷掺杂碳材料的比表面积随着磷酸用量的增加逐渐增大。这表明磷酸具有活化碳材料结构,调控碳材料比表面积的作用。
由图1还可以看出,没有经过磷酸改性的样品,其孔道集中分布在(0.4~0.5) nm,平均孔径为0.46 nm。经磷酸改性后,获得的P@C催化剂样品孔径进一步扩大。当磷酸用量为2.5 mL时,所制备的样品平均孔径为0.54 nm;进一步提高磷酸用量,样品的孔径进一步地扩大,P@C-5和P@C-10的平均孔径分别达到了0.65 nm和0.66 nm。这些样品的孔径不仅分布窄,而且孔径大小非常接近环己烷和苯分子的临界直径,这不仅有助于反应体系中物质在催化剂内外表面的扩散,同时可能存在的择形效应也将有助于抑制较大分子产物的生成,从而提高催化剂寿命。另一方面,P@C样品在(1.8~2.5)nm处出现少量的介孔,并且介孔孔径随磷酸用量增加而略有增加,这样有助于微孔孔道的贯通,更有益于物质的扩散。
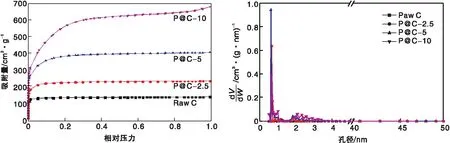
图1 不同催化剂的N2吸附-脱附等温曲线和孔径分布图Figure 1 N2 adsorption-desorption isotherms and pore size distribution of different catalysts

表1 不同催化剂的元素含量、比表面积、孔容和孔径数据
由表1各样品的孔容结果可知,Raw C的孔容为0.36 cm3·g-1,磷酸活化后的P@C-2.5、P@C-5、P@C-10样品,其孔容分别增加到0.62 cm3·g-1、0.75 cm3·g-1和1.03 cm3·g-1,表明随着磷酸添加量的增加,磷含量提升的同时碳材料的孔体积也随之增大。以上结果再次表明,磷酸加入对孔道结构起到了很好的调节修饰作用。通过控制磷酸添加量,可以方便地获得孔径分布集中、表面积大的微孔碳材料。
为了进一步探究磷酸加入量对碳材料结构的影响,对制备的样品进行XRD和Raman表征,结果见图2和图3。

图2 各催化剂样品的XRD图Figure 2 XRD patterns of catalysts
从图2可以看出,各样品在2θ为24°和44°处出现两个峰,分别对应石墨化结构的(002)和(100)晶面,表现出明显的无定形碳材料的结构特征。随着磷酸添加量的增加,两个峰均逐渐向小角度偏移,这表明磷酸的加入使得碳材料中晶面间距随之逐渐增大[10-11],说明P原子有效地掺入了碳材料骨架。

图3 各催化剂样品的拉曼谱图Figure 3 Raman spectra of catalysts
从图3可以看出,四种样品均在1 350 cm-1和1 590 cm-1处有特征峰,分别对应碳材料的D带与G带,与材料的无序度和石墨化程度紧密相关。D带与G带的强度比(ID/IG)用来表示碳材料的缺陷程度,ID/IG值越大,其缺陷越多,有序度越低[12-13]。计算得到Raw C、P@C-2.5、P@C-5、P@C-10的ID/IG值分别为0.936、0.961、0.997和1.052。样品的ID/IG值随着磷酸添加量增加而增大,表明随磷酸用量的增加掺入碳骨架中的磷原子也增加[14],从而导致碳骨架中引入了更多的缺陷。
2.2 磷酸用量对碳材料中磷物种存在形式的影响
为研究磷掺杂微孔碳材料P@C中磷掺杂量及其磷的化学状态,对制备的样品进行了XPS分析,结果如图4所示。
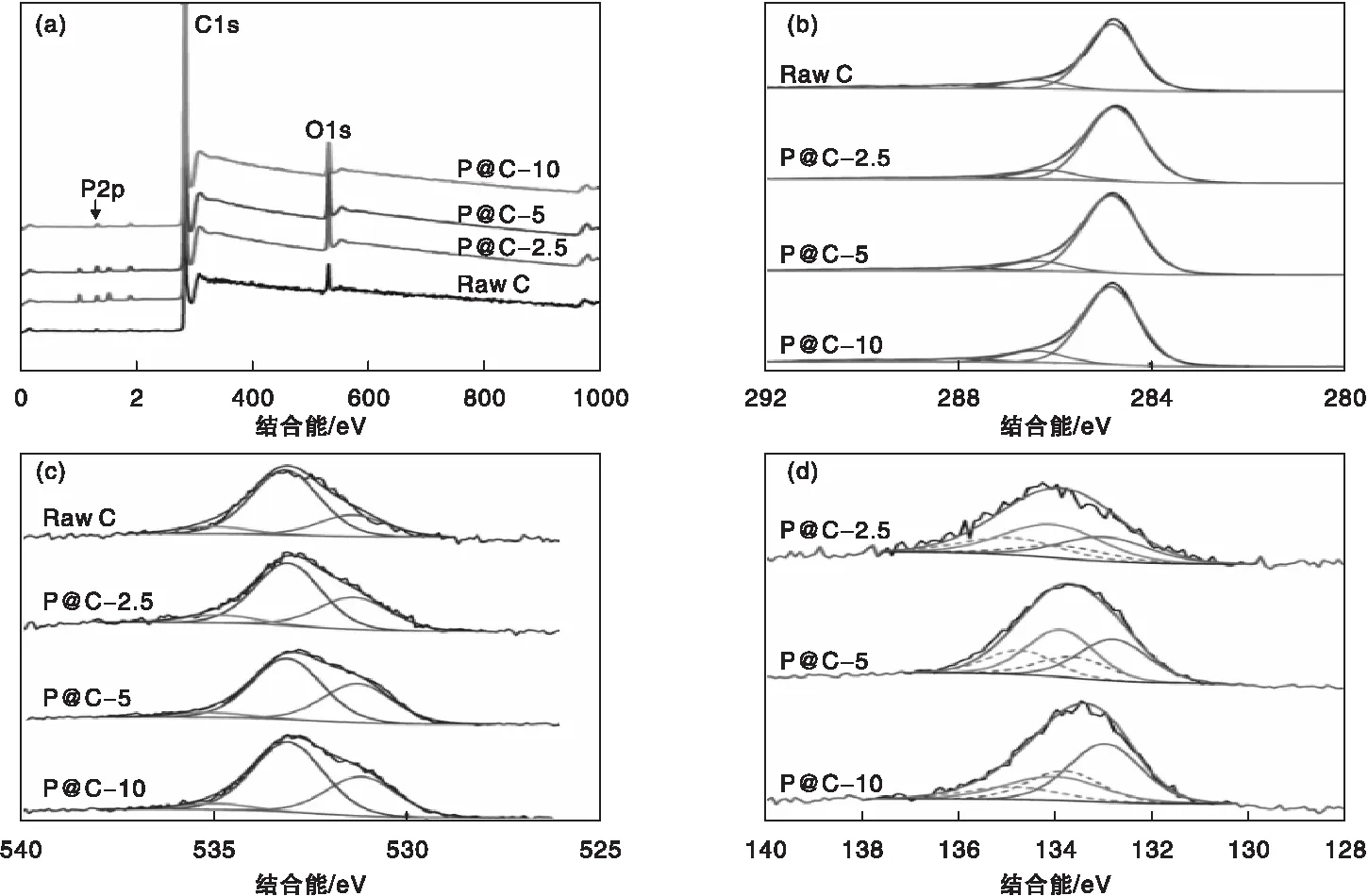
图4 催化剂的XPS能谱分析(a) XPS总谱;(b) C1s谱图;(c) O1s谱图;(d) P2p谱图Figure 4 XPS spectrum of catalysts(a) XPS survey;(b) high-resolution XPS C1s spectra; (C) XPS O2p spectra;(d) XPS P2p spectra.
由XPS总谱图以及表1可知,未经过磷酸改性的样品只含有C、O两种元素,含量分别为94.78%和5.22%,经过磷酸改性后的P@C催化剂中,均含有C、O、P三种元素。在P@C-2.5,P@C-5和P@C-10中,O元素含量分别为5.66%、5.92%和9.00%,P元素含量分别为1.08%、1.53%和2.03%。这表明随着制备过程中磷酸用量的增大,更多的P原子进入了碳骨架中,这与XRD和Raman表征结果一致。同时,随着P掺杂量的增加,P@C催化剂中O含量也随之增加。这是因为在P掺杂过程中通常会伴随氧元素的引入[15-17]。P@C-2.5、P@C-5和P@C-10中所含的P元素与O元素物质的量比分别为1∶5.24、1∶3.87和1∶4.33。这表明随着P含量的增加,C、O、P三元素组成的官能团可能有所不同。
对P@C-5和Raw C样品进行NH3-TPD吸脱附测试,结果如图5所示。由图5可见,不含磷的Raw C样品没有出现明显的NH3脱附峰,脱附曲线基本和基线相重合,这表明未经过磷掺杂改性的Raw C表面基本上不具有酸性中心。而P@C-5样品出现了显著的NH3脱附峰,酸中心温度约为173 ℃,经过积分计算酸量约为100 μmol·g-1。这表明经过磷酸处理后的样品上有大量的弱酸中心,是一种具有较高酸密度的弱酸性碳基材料。根据上述的XPS表征表明,这些弱酸中心应该是其表面丰富的C、P、O元素组成的官能团所引起的。
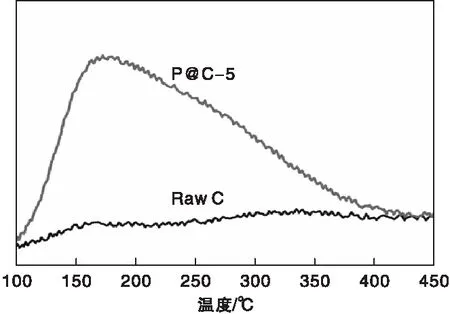
图5 Raw C和P@C-5样品的NH3-TPD 谱图Figure 5 NH3-TPD spectra of Raw C and P@C-5 sample
2.3 磷酸用量对环己烷脱氢反应的影响
将Raw C、P@C-2.5、P@C-5、P@C-10样品用于环己烷催化脱氢反应,在525 ℃下考察了制备过程中磷酸用量对催化剂催化性能的影响,结果如图6所示。由图6可知,以Raw C为催化剂,环己烷转化率仅为2.8%,同时苯选择性低于10%。表明尽管未掺杂的碳表面有丰富的含氧官能团,但这些官能团对环己烷转化基本惰性而且对苯的生成没有明显的选择性。经磷掺杂后,P@C催化剂的催化性能得到显著提高。在P@C-2.5催化剂作用下,环己烷转化率升高到70.2%,同时苯选择性达到98%以上,产氢速率0.319 mmol·(g催化剂·min)-1。这表明P@C催化剂对环己烷脱氢反应有很好的催化活性,但难以催化环己烷开环反应。NH3-TPD吸附-脱附测试结果说明,磷掺杂过程中在碳基样品表面引入了大量的弱酸中心,而这些弱酸中心可以选择性地催化环己烷脱氢而不能催化开环反应,从而对环己烷脱氢生成苯的反应具有十分优良的选择性。随着磷酸用量的增大,环己烷转化率进一步提高,P@C-5作为催化剂时,环己烷转化率达到89.1%,产氢速率达0.413 mmol·(g催化剂·min)-1,苯选择性达到100%,色谱条件下检测不到苯以外的其他有机产物。继续增加磷酸用量,以P@C-10为催化剂时,环己烷转化率反而降低到84.1%,产氢速率降为0.380 mmol·(g催化剂·min)-1,且苯选择性降低到97.5%左右。这可能是由于磷酸用量的增加,P的物种形态发生了变化导致的。

图6 磷酸用量对环己烷转化率和苯选择性的影响Figure 6 Effect of phosphoric acid amount on conversion of n-hexane and selectivity of benzene
2.4 反应温度对环己烷脱氢反应的影响
采用活性更优的P@C-5为催化剂,进一步考察反应温度对环己烷脱氢反应的影响,结果如图7所示。由图7可知,反应温度为480 ℃时,环己烷转化率为40%,苯选择性将近100%。随着反应温度的升高,环己烷转化率不断升高。这是由于环己烷脱氢是一个强吸热反应,升温有利于环己烷的转化。当反应温度提高至550 ℃,环己烷转化率虽然将近100%,但苯的初始选择性下降到91%左右,且随着反应时间的延长,苯选择性进一步下降。气相色谱分析表明此时的副产物为低碳烷烃和烯烃。这也表明高温促进了环己烷的开环裂解。从以上分析可以看出,当反应温度为525 ℃时,不仅环己烷转化率和苯选择性高,且催化剂性能稳定,连续反应400 min后,环己烷转化率和苯选择性未出现明显下降,产氢速率保持0.41 mmol·(g催化剂·min)-1左右,说明催化剂在此条件下具有良好的催化脱氢活性、优异的苯选择性和稳定性。这将给环烷烃脱氢催化剂设计提供了新的思路[21-24],也为有机液体储氢体系的应用提供了一个可能的选项。
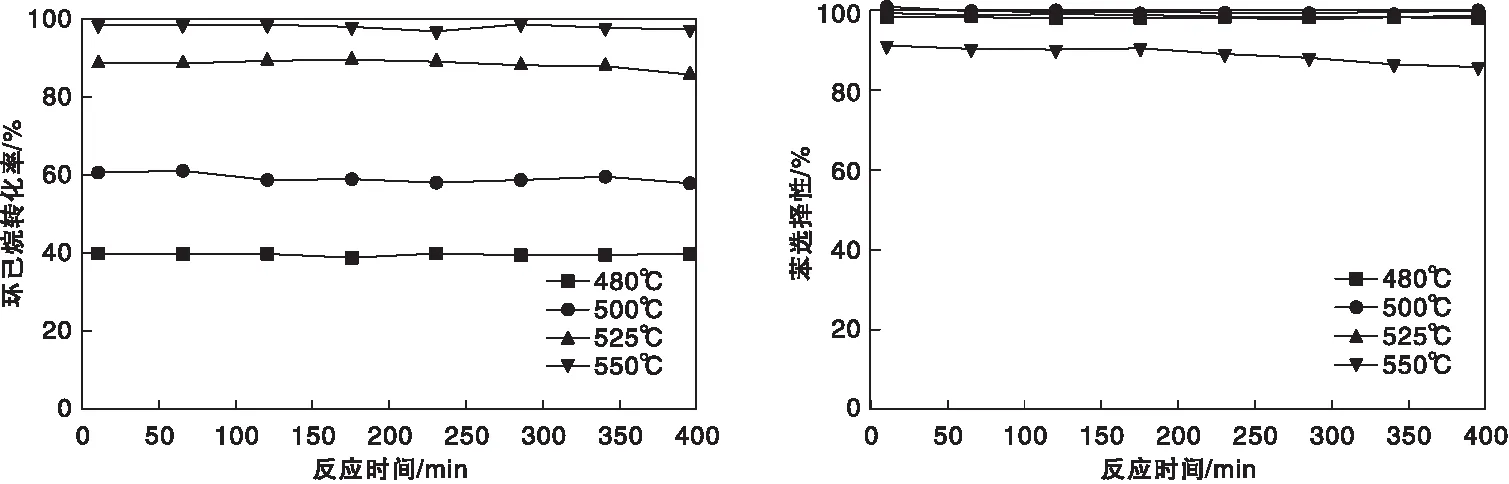
图7 反应温度对环己烷转化率和苯选择性的影响Figure 7 Effect of reaction temperature on conversion of cyclohexane and selectivity of benzene
3 结 论
采用淀粉为碳源、磷酸为磷源制备了孔径分布窄、比表面积大的微孔磷掺杂碳材料P@C。磷酸在制备过程中不仅作为磷源,还起到活化剂的作用,参与调控催化剂的比表面积、孔容和孔径的大小。磷掺杂的过程中伴随着氧的引入,在碳基材料的表面形成了大量的弱酸性的C、P、O元素组成的官能团,这些酸性官能团可以作为催化活性中心,对环己烷脱氢制苯反应具有优良的催化活性和选择性。同时P掺杂过程中所产生丰富的孔道结构和合适的孔径分布也十分有益于反应的进行。总之,P@C催化剂成本低、合成方便、对环己烷脱氢制苯反应活性高、性能稳定。将给环烷烃脱氢催化剂设计提供了新的思路,也为有机液体储氢体系的应用提供了一个可能的选项。
