2013~2019年青岛市新生儿高苯丙氨酸血症筛查情况及基因突变分析
2020-08-26吕金峰候方王伟青陈艳萍
吕金峰,候方,王伟青,陈艳萍
1青岛市妇女儿童医院,山东青岛266034;2青岛大学附属医院
高苯丙氨酸血症(HPA)是一种常染色体隐性遗传性代谢病,是我国最早进行的新生儿疾病筛查病种之一。HPA根据病因可分为苯丙氨酸羟化酶(PAH)缺乏症及其辅酶四氢生物蝶呤(BH4)缺乏症两类。HPA患儿因酶的缺乏,体内的苯丙氨酸(Phe)不能代谢为酪氨酸(Tyr),Phe及其旁路代谢产物在体内蓄积而损伤神经系统,导致智力发育落后、小头畸形、癫痫、自闭等。早筛查、早诊断、早治疗可以避免HPA患儿发生神经系统损伤,保障患儿的智力和体格发育水平。随着国家对新生儿出生缺陷的重视,该病的筛查覆盖率越来越广。本研究对青岛市新生儿HPA筛查情况进行调查,并对HPA患儿进行基因变异分析。现报告如下。
1 资料与方法
1.1 临床资料 选择2013年1月1日~2019年12月31日在青岛市具有接产资格的96家医院出生并参加HPA筛查的新生儿375 697例,均出生满72 h且充分哺乳6次以上,其中男195 242例、女180 455例。本研究通过医院伦理委员会审核,新生儿监护人均签署知情同意书。
1.2 HPA筛查方法 采用串联质谱非衍生化法。采集新生儿足后跟血滴于Whatman903滤纸片上,血斑数3~4个、直径>8 mm,自然阴干。排除存在溶血、反复挤压、重复滴血、污染等情况的不合格样本。采用液相色谱-质谱联用分析仪TQ Detector(美国Waters公司)检测血浆Phe、Tyr,严格按照非衍生化法多种氨基酸、肉碱和琥珀酰丙酮测定试剂盒(串联质谱法,美国PerkinElmer公司)说明书进行操作。根据2014年的HPA诊治共识[1],串联质谱法检测Phe>120 μmol/L且Phe/Tyr>2.0则确诊为HPA。对确诊HPA的患儿行检尿蝶呤谱分析、红细胞DHPR活性测定、BH4负荷试验及基因检测,区分PAH缺乏症和BH4缺乏症。根据血浆Phe水平评价PAH缺乏症:Phe为120~360 μmol/L定义为轻度HPA,360~1 200 μmol/L定义为轻度苯丙酮尿症(PKU),≥1 200 μmol/L定义为经典PKU。
1.3 基因突变位点检测方法 采用高通量测序法。对于确诊HPA的患儿,抽取患儿及其父母外周血2 mL,使用QIAGEN试剂盒,按照Illumina标准化流程制备基因组DNA。纯化扩增后,对HPA患儿PAH及BH4合成代谢相关基因(PTS、GCH1)的184个突变位点的外显子及其侧翼序列进行高通量测序,采用Sequencing、GeneTool等软件对测序结果进行序列比对和分析,对患儿父母采用Sanger测序进行变异基因的来源验证。所有基因突变位点经过PAH数据库和PubMed数据库确定突变类型、核苷酸突变、蛋白质改变、突变频数、突变频率及其相关表型。
1.4 统计学方法 采用SPSS20.0统计软件。计数资料以例表示,结果比较采用χ2检验。P<0.05为差异有统计学意义。
2 结果
2.1 HPA筛查结果 参加HPA筛查的375 697例新生儿中,确诊HPA 69例,PAH缺乏症64例、BH4缺乏症5例,PAH缺乏症患儿中轻度HPA 23例、轻度PKU 22例、经典PKU 19例。2013~2019年HPA发病率比较P均>0.05,平均发病率为1.84/万。见表1。
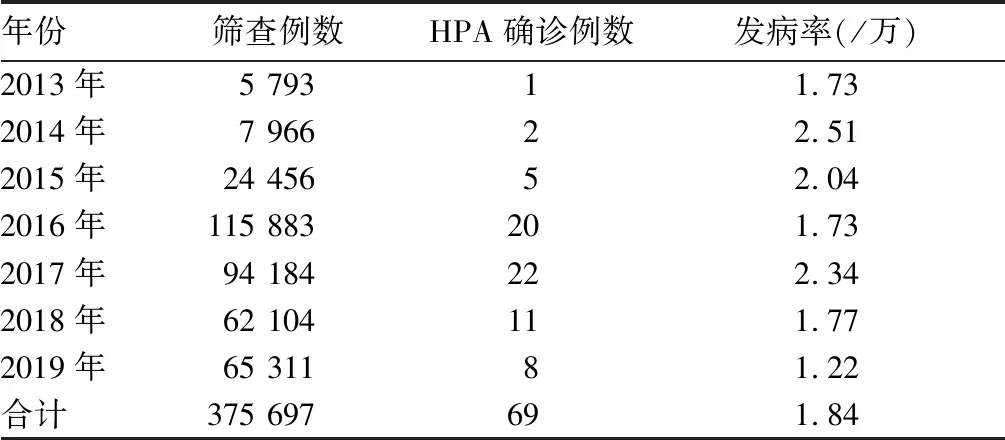
表1 青岛地区2013~2019年新生儿HPA筛查结果
2.2 基因突变位点检测结果 54例HPA患儿行基因突变位点检测,其中纯合突变1例、杂合突变7例、复合杂合突变46例,存在PAH基因突变50例、BH4基因(PTS基因)突变4例。50例PAH基因突变患儿突变情况及其相关表型见表2。
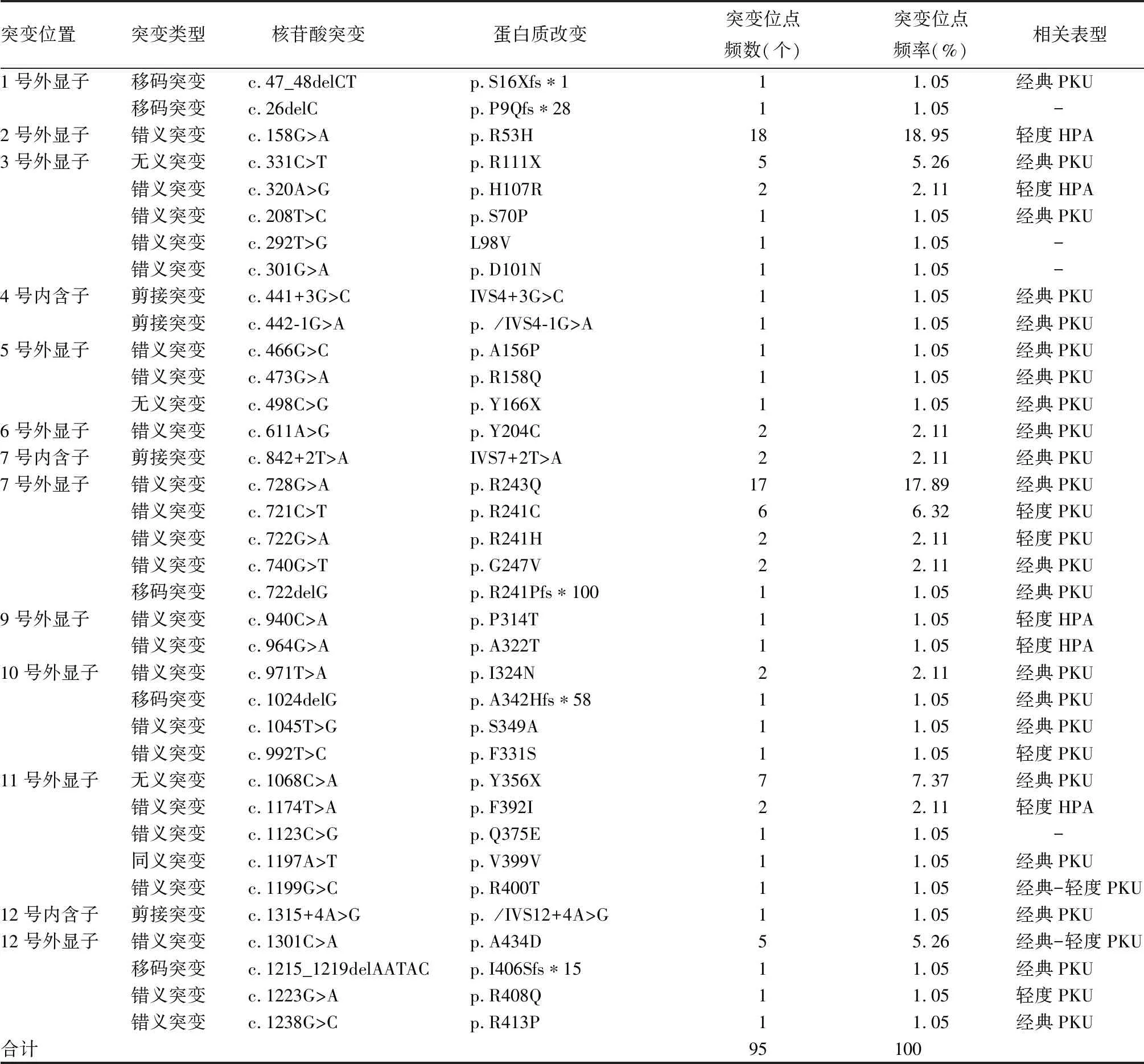
表2 50例PAH基因突变患儿突变情况及其相关表型
3 讨论
HPA是一种常染色体隐性遗传病,表现为体内的Phe不能代谢为Tyr。Phe的正常代谢需要PAH和BH4的共同参与,PAH基因突变可导致患者体内PAH缺乏,BH4合成代谢中相关基因(PTS、GCH1、SPR、QDPR和PCBD1)突变可导致患者体内BH4缺乏。近年来研究发现,DNAJC12基因突变也可以导致人体内PAH酶活性降低[2~4]。PAH活性降低或缺乏可导致Phe羟化受阻,Phe在体内大量蓄积,引起智力发育落后、抽搐、小头畸形、毛发颜色浅等。同时,由于旁路代谢途径的增强,大量的苯乳酸从尿中排出,使患儿尿液具有特殊的鼠尿味。BH4是PAH、色氨酸羟化酶和酪氨酸羟化酶的辅酶,其缺乏可导致肌张力低下、运动障碍、嗜睡、反应迟钝、小脑发育障碍等严重的神经系统症状。HPA的发病率具有明显的地域差异,土耳其发病率为1∶2 600[5],欧洲为1∶8 000[6],美国为1∶15 000[7],韩国为1∶41 000[8],而中国的总体发病率为1∶10 397[1]。本研究中青岛市7年平均发病率为1∶5 445(1.84/万),高于中国总体发病率。
本研究对54例HPA患儿进行了基因检测,其中50例存在PAH基因变异,共95个变异等位基因,检出率达95%,与宋昉等[9]报道的北方地区平均检出率(94.6%)接近。本研究共检测到36个不同的变异位点,最常见的变异位点为p.R53H(18.95%)、p.R243Q(17.89%)、p.Y356X(7.37%)、p.R241C(6.32%)、p.A434D(5.26%)、p.R111X(5.26%),与陈晨等[10]、罗静思等[11]报道的变异位点基本相同,但未检测到河南和福建地区常见的突变EX6-96A>G[10,12],仅检测到1次广西地区[11]最常见的突变p.R408Q。本研究基因检测结果与王慧琴等[13]报道的新疆维吾尔族HPA突变类型差异很大,因此不同地区的HPA突变数据不尽相同。
本次筛查的患儿中PAH基因存在等位基因异质性,最常见的变异类型为错义突变(74.74%),其次为无义突变(13.68%)。对PAH数据库(http://biopku.org/home/pah.asp)进行检索,截止2019年11月10日收录的PAH基因突变数量为1 184 个,变异位点主要位于7号外显子、6号外显子、3号外显子和11号外显子。本研究中PAH基因变异频率最高的也是7号外显子(29.47%),其次为2号外显子(18.95%)和11号外显子(12.63%)。本研究50例PAH缺乏症患儿中,7例杂合突变患儿通过临床表现确诊,2例确诊为轻度PKU,5例确诊为轻度HPA。这7例患儿的基因检测结果与该病的遗传规律不相符,可能的原因有:①受该基因检测技术的限制,只能检测已知的变异位点,不能检测新的突变位点及大片段缺失或重复。②未进行相关突变基因DNAJC12[3~5]的检测。③存在其他影响蛋白质功能的因素等。另外,本研究4例患儿确诊为BH4缺乏症,携带有PTS基因,为6-丙酮酰四氢蝶呤合成酶缺乏型。
综上所述,2013~2019年青岛市新生儿HPA平均发病率高于全国水平,PAH基因最常见的变异位点为p.R53H、p.R243Q,并存在等位基因异质性。基因检测不仅能鉴别HPA的类型,还可通过基因型和表型的关系进行疾病的分型、预后判断,并对临床生化表型不典型者进行确诊。对已经明确基因突变的患儿进行家系分析和遗传咨询,有利于提高出生人口素质。但基因检测操作复杂、技术具有局限性、检测周期长、价格昂贵,不适用于大规模的新生儿疾病筛查。
