芪精镇痛颗粒质量标准研究
2020-08-22张秋红贾庆文
张秋红,贾庆文
(1. 济南市食品药品检验检测中心,山东 济南 250102;2. 山东福瑞达医药集团有限公司,山东 济南250101)
芪精镇痛颗粒,是依据山东已故中医药名家周凤梧教授拟定的芪精镇痛汤[1]治疗气血亏虚型头痛的经验组方研制而成的制剂,目前为济南某医疗机构自制制剂,由炙黄芪、当归、茯苓、炙甘草、大枣、川芎、升麻等十味中药组成。经多年临床使用证明,该制剂在补气养血、疏风散痛等方面有较好疗效,临床上主要用于气血亏虚型头痛。原制剂质量标准简单,仅有当归、川芎的薄层色谱(TLC)鉴别和常规制剂通则项下的检查项目,缺少君药与其他药味的鉴别和含量测定,无法较全面控制制剂质量。为保证芪精镇痛颗粒质量满足疗效,同时符合山东省医疗机构制剂标准提高的要求,本实验从定性定量两个方面对该制剂进行全面研究,以期达到控制其质量的目的。
1 仪器与试药
1.1 仪器
LC-20A高效液相色谱仪,SPD-20A检测器(日本岛津); AE-100型电子天平(Mettler);AE-240电子天平(Mettler);WINCATS薄层照相系统(CAMAG);硅胶G薄层板(Merck);定量毛细管(美国Drummond公司);HH-4数显恒温水浴锅(上海科恒)。
1.2 试药
黄芪甲苷对照品(批号:110781-201717),黄芪对照药材(批号:121462- 201705),升麻对照药材(批号:121182-201102),甘草对照药材(批号:120904- 201620),阿魏酸对照品(批号:110773-201915),异阿魏酸对照品(批号:111698-201904)均购自中国食品药品检定研究院。芪精镇痛颗粒(济南某医疗机构自制制剂,批号:20190108,20190110,20190201,20190210,20190301,20190310,20190401,20190410,20190501,20190510)。甲醇、乙腈为色谱纯,水为纯净水,其余试剂均为分析纯。
2 方法与结果
2.1 TLC鉴别[2]
2.1.1 炙黄芪[3]取本品颗粒3 g,加水5 ml使浸润,加入用水饱和的正丁醇30 ml,摇匀,超声10 min,滤过,滤液用氨试液洗涤2次,每次25 ml,取正丁醇液,蒸干,加甲醇1 ml溶解残渣,作为样品溶液。另取黄芪对照药材1 g,同法制成黄芪对照药材溶液。再取黄芪甲苷对照品,加甲醇制成每1 ml含1 mg的黄芪甲苷对照品液。取去除炙黄芪制得的芪精镇痛颗粒,按同法制成缺黄芪的阴性样品溶液。吸取上述黄芪甲苷对照品液2 µl,其余3种溶液各10 µl,分别点于同一硅胶G薄层板上,以三氯甲烷-甲醇-水(13:7:2)下层液为展开剂,展开,取出,晾干,喷硫酸乙醇溶液(1→10),105 ℃加热显色。供试品色谱中,与对照品、对照药材相应位置上,显棕色至棕褐色斑点;紫外光(365 nm)下显橙黄色斑点。缺炙黄芪的阴性样品无干扰,结果见图1~2。
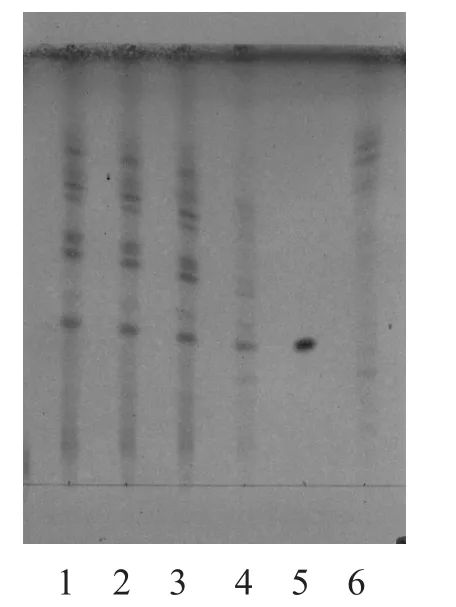
图1 炙黄芪的TLC图(日光下)
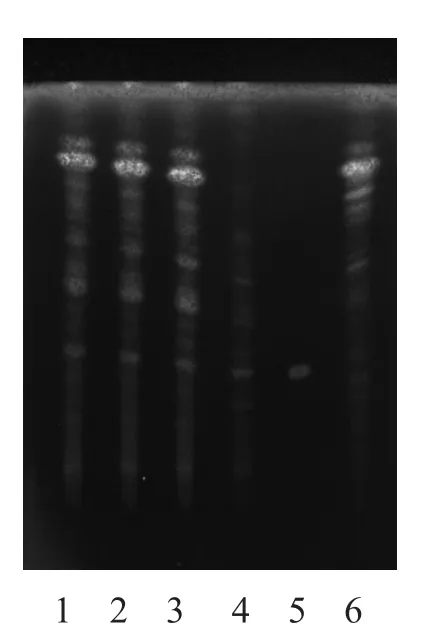
图2 炙黄芪的TLC 图(365 nm)
2.1.2 升麻[3-4]取本品颗粒3 g,加乙醇30 ml,水浴回流1 h,滤过,蒸干滤液,加乙醇1 ml溶解残渣,作为样品溶液。另取升麻对照药材1 g,同法制成升麻对照药材溶液。再取异阿魏酸对照品,制成每1 ml含1 mg的乙醇溶液,作为异阿魏酸对照品液。取去除升麻制得的芪精镇痛颗粒,按同法制成缺升麻的阴性样品溶液。吸取上述样品溶液15 µl、对照药材溶液、对照品液各2 µl、缺升麻的阴性样品溶液10 µl,分别点于同一硅胶G薄层板上,以甲苯-三氯甲烷-冰醋酸(12:2:1)为展开剂,展开,取出,晾干,紫外光(365,254 nm)下检视。供试品色谱中,在与对照药材和对照品色谱相应的位置上,显相同的蓝色荧光斑点。缺升麻的阴性样品无干扰,结果见图3~4。
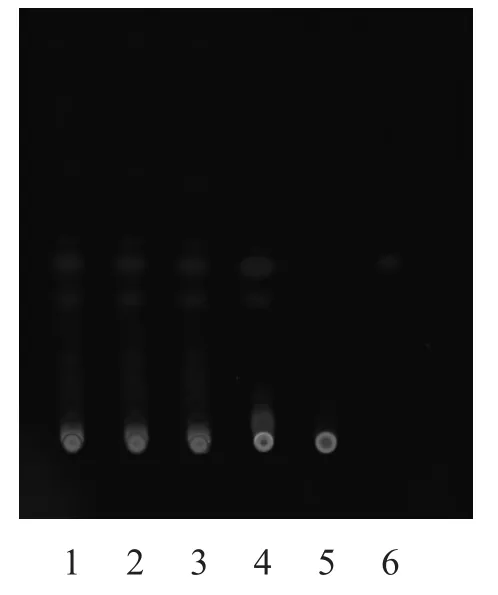
图3 升麻的TLC 图(365 nm)
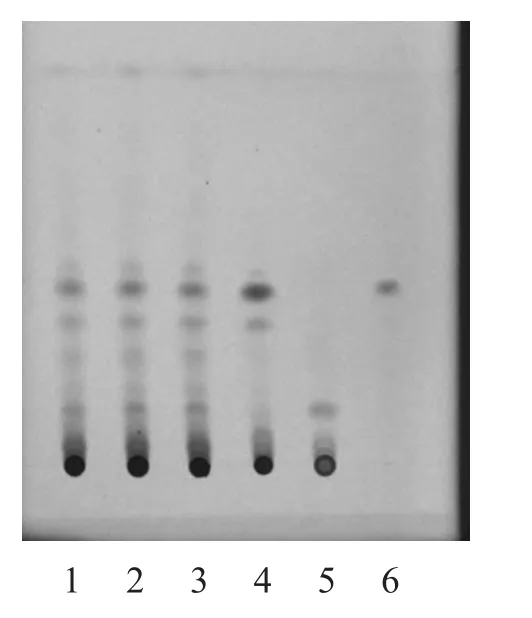
图4 升麻的TLC图(254 nm)
2.1.3 炙甘草[3]取本品颗粒3 g,加乙醚40 ml,回流1 h,滤过,弃去乙醚液,残渣加甲醇30 ml,回流1 h,滤过,滤液蒸干,残渣加水40 ml使溶解,再用水饱和的正丁醇20 ml萃取3次,合并正丁醇液,用正丁醇饱和的水洗涤3次,取正丁醇液蒸干,加甲醇5 ml溶解残渣,作为样品溶液。另取甘草对照药材1 g,同法制成甘草对照药材溶液。取去除炙甘草制得的芪精镇痛颗粒,按同法制成缺炙甘草的阴性样品溶液。吸取上述3种溶液各5 µl,分别点于同一用1 % NaOH制备的硅胶G薄层板上,以乙酸乙酯-甲酸-冰醋酸-水(15:1:1:2)为展开剂,展开,取出,晾干。喷硫酸乙醇(1→10)液,105 ℃加热至斑点清晰。供试品色谱中,在对照药材色谱相应的位置上,显相同的3个黄色斑点;紫外光(365 nm)下检视,显相同的6个荧光斑点。缺炙甘草的阴性样品无干扰,结果见图5~6。
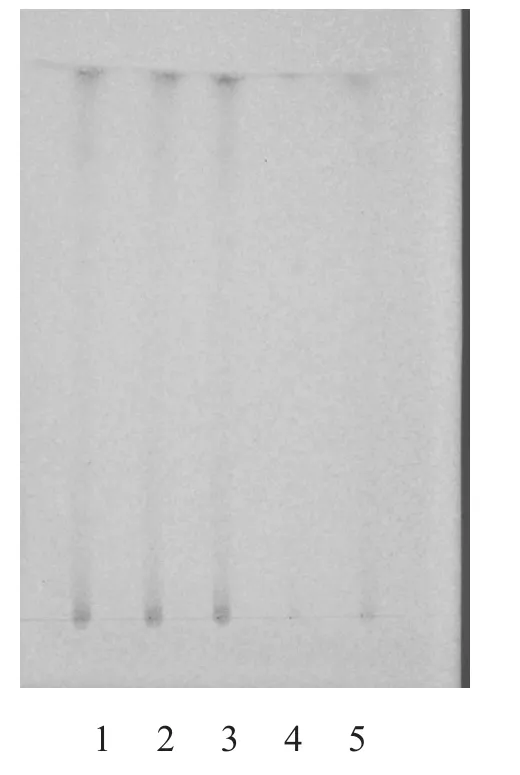
图5 炙甘草的TLC 图(日光)
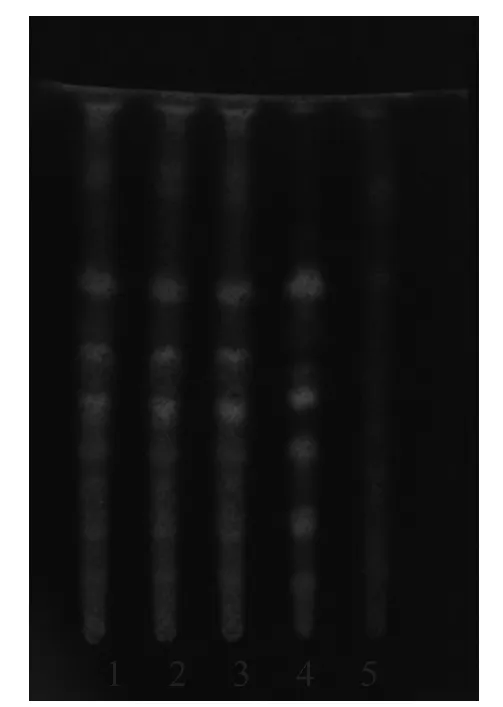
图6 炙甘草的TLC图(365 nm)
2.2 HPLC含量测定[2]
2.2.1 色谱条件 岛津 Wondasil C18色谱柱(250 mm×4.6 mm,5 µm);流动相乙腈-0.1 %磷酸溶液(1:9);检测波长321 nm;流速1.0 ml/min;柱温30 ℃;进样量10 µl。理论板数按阿魏酸计应不低于4800。
2.2.2 溶液的制备
2.2.2.1 对照品溶液制备 精密称取阿魏酸对照品、异阿魏酸对照品适量,加70 %甲醇制成每1 ml含阿魏酸、异阿魏酸各20 µg的混合溶液,即得。
2.2.2.2 供试品溶液的制备 取芪精镇痛颗粒细粉约1 g,精密称定,置具塞锥形瓶中,精密加入70 %甲醇25 ml,密塞,称定重量,回流30 min,放冷,用70 %甲醇补足减失的重量,摇匀,滤过,取续滤液,即得。
2.2.2.3 阴性样品溶液的制备 处方中去除当归、川芎、升麻、酸枣仁后制成芪精镇痛颗粒含量测定用阴性样品, 并按2.2.2.2项制备方法制成含量测定用阴性样品溶液。
2.2.3 专属性试验 分别吸取上述3种溶液各10 µl,注入液相色谱仪,混合对照品、供试品及阴性样品色谱见图色谱图见图7~9。表明该方法专属性良好。
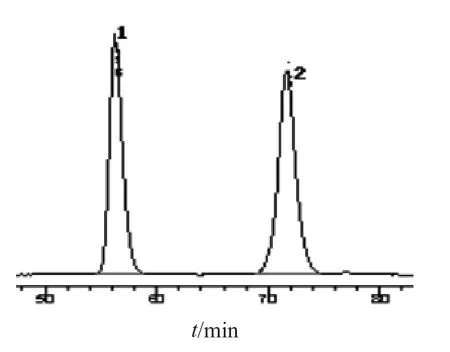
图7 对照品溶液的HPLC图
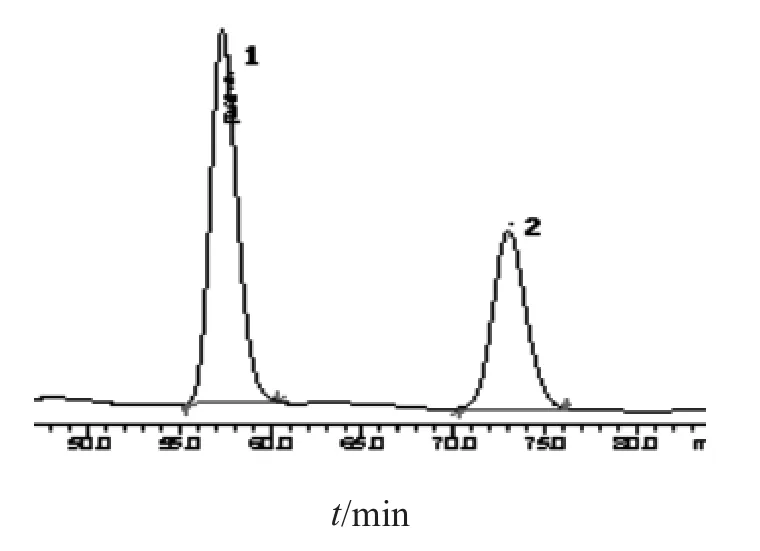
图8 芪精镇痛颗粒样品溶液的HPLC图
图9 芪精镇痛颗粒含量测定阴性样品溶液的HPLC图
2.2.4 线性关系考察 精密吸取阿魏酸、异阿魏酸混合对照品溶液(阿魏酸20.2 µg/ml,异阿魏酸20.4 µg/ml)2,5,10,20,40 µl,按2.1.1项下方法分别注入液相色谱仪,测定峰面积。分别以阿魏酸进样量(µg)为横坐标,峰面积为纵坐标;以异阿魏酸进样量(µg)为横坐标,峰面积为纵坐标,绘得标准曲线。阿魏酸的回归曲线方程为Y=5.31×103X+7.90×102,r=1.0000;异阿魏酸的回归曲线方程为Y=4.84×103X+1.71×103,r=1.0000。结果表明,阿魏酸在40.4~808 ng(r= 1.0000)范围内线性关系良好,异阿魏酸在40.8~816 ng(r=1.0000)范围内线性关系良好。
2.2.5 稳定性试验 精密吸取批号为20190510的供试品溶液10 µl,室温下分别于0,2,6,12,18,24 h,注入色谱仪,测定阿魏酸、异阿魏酸峰面积,计算RSD,分别为1.09 %(n=6),1.67 %(n=6)。表明供试品溶液在24 h内稳定性良好,不影响阿魏酸与异阿魏酸成分的测定。
2.2.6 精密度试验 精密吸取对照品溶液10 µl,注入色谱仪,测定阿魏酸异阿魏酸峰面积,连续测定6次,计算RSD,分别为0.17 %(n=6),1.28 %(n=6)。表明仪器的精密度良好。
2.2.7 重复性试验 称取同一样品(批号20190201)6份,每份约1 g,精密称定,按2.2.2.2项方法制成供试品溶液,每份精密吸取10 µl,注入液相色谱仪,测定阿魏酸、异阿魏酸的含量,计算RSD,结果分别为1.00 %(n=6),1.85 %(n=6)。表明方法的重复性良好。
2.2.8 加样回收试验 取含阿魏酸0.4683 mg/ml,含异阿魏酸0.2504 mg/ml的芪精镇痛颗粒(批号20190201)12份,研细,6份精密称取约0.5 g加入阿魏酸对照品溶液(20.2 µg/ml)10 ml,6份加入异阿魏酸对照品溶液(20.76 µg/ml)10 ml。按2.2.2.2项方法制成不同的12份供试品溶液,按2.2.1项色谱条件,注入液相色谱仪,测定阿魏酸、异阿魏酸峰面积,计算回收率。两个成分的RSD均未超过2.0 %,表明该方法准确性良好。见表1。
2.2.9 样品含量测定 称取样品粉末各约1 g,精密称定,制成供试品溶液,分别精密吸取10 µl,注入色谱仪,测定阿魏酸、异阿魏酸含量,结果见表2。
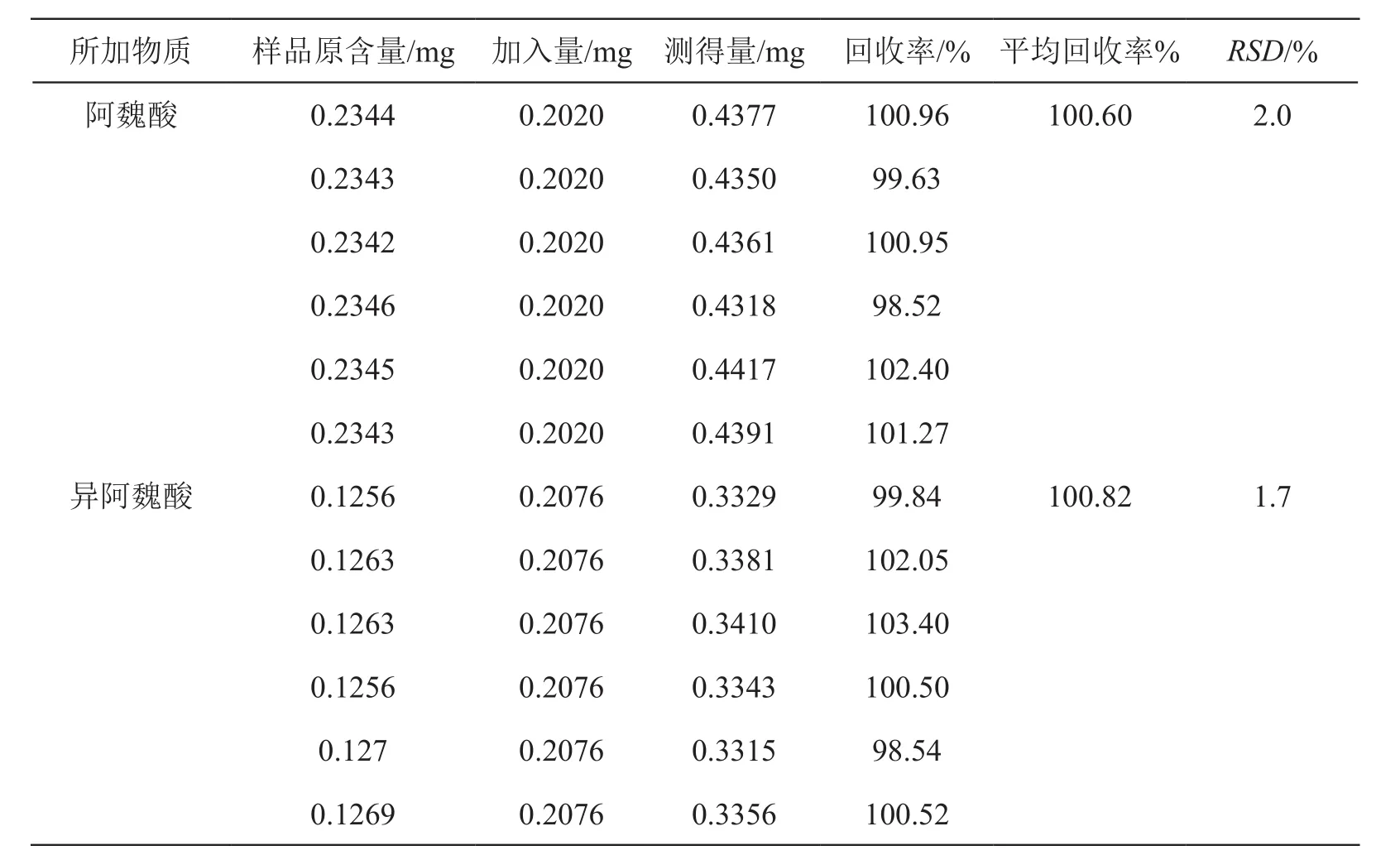
表1 加样回收试验结果
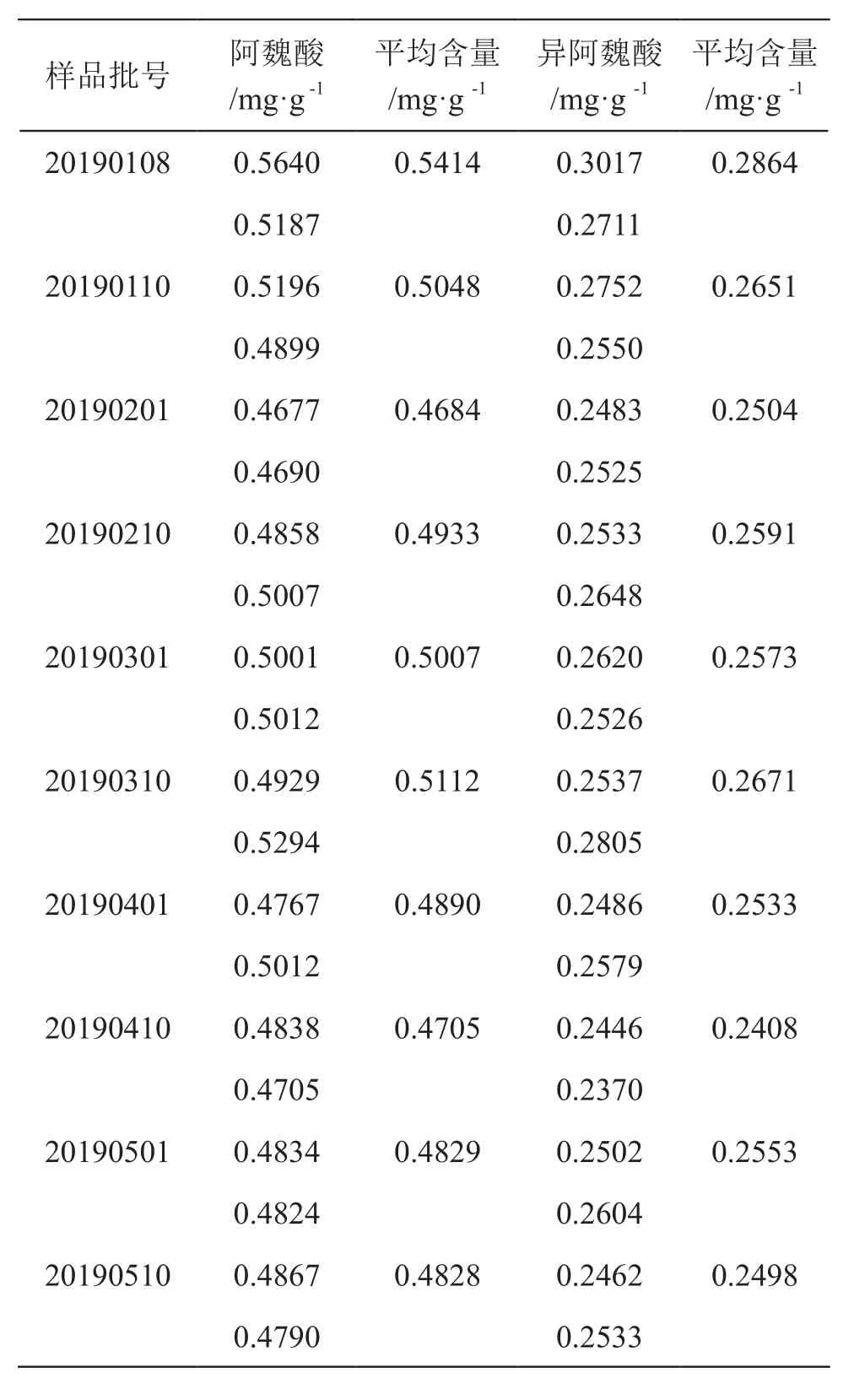
表2 芪精镇痛颗粒中阿魏酸、异阿魏酸含量
10批样品含量测定结果均大于0.75 mg/g,根据测定结果规定本品每1 g含阿魏酸(C21H20O9)与异阿魏酸(C21H20O9)的总量不得少于0.45 mg。
3 讨论
3.1 TLC鉴别的讨论
3.1.1 炙黄芪 3种提取方法均可用于炙黄芪定性,但用超声法提取的供试品溶液杂质多,底色较深,且原点附近有黑色斑点拖尾严重;用中性氧化铝柱提取的供试液,能有效除去糖类及脂溶性的干扰。但由于方中所含药物较多,成分复杂,故无效斑点亦较多。而用碱性溶剂(氨水)提取的供试品溶液能有效去除阿魏酸[5]、异阿魏酸、甘草酸等酸性成分,氨水洗涤的另一重要作用是将其他黄芪皂苷类成分转化为黄芪甲苷,提高黄芪甲苷的含量,有利于测定。结果表明,色谱分离效果更好,色谱斑点清晰,无干扰,黄芪甲苷斑点的Rf值约为0.42~0.52。综合考虑,用氨水洗涤法是最简便可行的提取方法。但预饱和后仍有较为明显的边缘效应,需进一步查找原因找出可行方法减少边缘效应影响。
3.1.2 升麻 在此实验中,以苯-三氯甲烷-冰醋酸(12:2:1)为展开剂进行展开,但在薄层板上出现展开剂前沿分层,考虑到所购的预制板结构不稳定,会影响展开效果;且展开剂本身极性不同,存在分层。综合考虑,将预制板先放入展开剂中空跑一遍,晾干,再作为展开板使用,效果可靠、明显。结果表明,经筛选优化后的条件,用于升麻的TLC鉴别,其分离效果好,斑点边缘清晰、圆整,不扩散,无拖尾,无边缘效应,可用于芪精阵痛颗粒中升麻的TLC定性鉴别分析。
3.1.3 炙甘草 炙甘草的TLC图谱显示,处方中其他成分及辅料均无干扰。该方法专属性强,重现性好,灵敏度高,且阴性无干扰,可用于芪精镇痛颗粒的质量控制。
3.2 HPLC含量测定的讨论
3.2.1 含量测定中供试品溶液的制备 分别采用70 %甲醇溶液、70 %乙醇溶液、50 %乙醇溶液、10 %乙醇溶液,10 %乙醇和70 %甲醇制备供试品溶液,测得含量相差不大,但过滤困难。以50 %乙醇、70 %乙醇溶液和70 %甲醇溶液为溶剂制备供试品溶液,测得含量差异较大,且50 %乙醇溶液过滤困难。综合考虑,最终选用70 %甲醇作为供试品溶剂。
3.2.2 阴性对照品制备 参照文献[5-8],当归、川芎、升麻、酸枣仁含所测成分,故选用剩余五味中药按处方工艺制备阴性对照品。
3.2.3 检测波长选择 根据阿魏、异阿魏酸在70 %甲醇溶液中的紫外吸相关文献[4,9-17],选择321 nm为检测波长。
3.2.4 流动相的选择 首先选用甲醇-1 %醋酸溶液(30:70),但阿魏酸、异阿魏酸达不到分离效果。后改变甲醇-1 %醋酸溶液比例(13:87),异阿魏酸分离效果不好,后选用乙腈-0.1 %磷酸溶液(13:87)作为流动相,异阿魏酸分离效果不好,后选用乙腈-0.1 %磷酸溶液(13:87),结果表明,阿魏酸、异阿魏酸分离效果良好,故选用乙腈-0.1 %磷酸溶液(13:87)为流动相。
3.2.5 含量测定结果分析 本实验建立了HPLC同时测定2种成分阿魏酸、异阿魏酸的含量,该方法精密度良好,灵敏度高,专属性好,可用于芪精镇痛颗粒中阿魏酸、异阿魏酸的含量测定。
综上,建立的质量标准方法简便可行,使芪精镇痛颗粒的质量得到有效控制。
