导致原发性Ⅰ型肾小管酸中毒的SLC4A1突变R388C致病机制的细胞学研究
2020-08-11潘鑫李亚彩王晓黎
潘鑫,李亚彩,王晓黎
(中国医科大学附属第一医院内分泌科,内分泌研究所,辽宁省内分泌疾病重点实验室,沈阳 110001)
Ⅰ型肾小管酸中毒又被称为远端肾小管酸中毒 (distal renal tubular acidosis,dRTA),在成人中常继发于其他疾病,如慢性肝脏疾病、自身免疫性疾病、肾移植、药物性肾损伤等。原发性dRTA多见于儿童,致病基因主要为编码位于顶膜H+-ATP酶B1和A4亚单位的ATP6V1B1(MIM:192132) 基 因和ATP6V0A4(MIM:605239) 基因,以及编码基底膜阴离子 (Cl-/HCO3-) 转运体1 (anion exchanger 1,AE1) 的SLC4A1(MIM:109270) 基因[1-2],其他基因还包括FOXI1(MIM:601093) 基 因[3]和WDR72(MIM:613214) 基因[4],但较为少见,除此之外还有15%左右的基因位点不明[2,5]。典型dRTA的临床表现为慢性代谢性酸中毒、低钾血症、高尿钙、肾脏钙质沉着和肾结石,尿液偏碱性。但原发性dRTA的临床表现异质性较强,部分不典型dRTA患者有可能至成年后才得到确诊,容易延误诊治[6-7]。本文对我院诊断为dRTA的1例成人患者及其父亲携带的一处新型SLC4A1基因杂合性突变R388C进行了细胞学研究,证实了该突变是导致dRTA的致病原因,并总结了目前已知的可导致dRTA的SLC4A1基因突变的类型和特点。
1 材料与方法
1.1 研究对象
研究对象为2018年在我院因低钾血症就诊、经基因诊断为原发性dRTA的1例患者及其父亲,2人均接受了病史采集、甲状腺功能、肾功能、尿常规、甲状腺和肾上腺相关激素以及影像学检查等,明确低钾血症的病因,并进行了二代基因测序。本研究已通过我院伦理委员会审核批准,所有研究对象均签署了知情同意书。
1.2 检测方法
采用全自动生化仪检测肝肾功能、血离子,采用免疫化学发光法测定促肾上腺皮质激素、皮质醇等。采用放射免疫法测定肾素、血管紧张素、醛固酮、胰岛素等。
提取患者及其父亲基因组DNA,利用经过生物素标记的探针 (P039-Exome,北京迈基诺公司) 与文库DNA杂交,进行外显子捕获,在NextSeq 500 (美国Illumina公司) 上进行高通量测序,测序数据经Burrows-Wheeler Aligner (v.0.7.10-r1039) 软件将其比对到人类基因组并分析 (基因组版本为GRCh37/hg19)。重点筛查与低钾血症临床表型相关的基因,并对检出的基因突变位点在患者及其父亲中采用一代测序验证。
1.3 细胞学实验
使 用GV230 (CMV-MCS-EGFP-SV40-Neomycin,吉凯基因) 质粒,利用XhoⅠ/KpnⅠ酶切位点导入野生型SLC4A1基因序列 (NM_000342) 和其突变型 (R388C) 基因序列,测序验证后,抽提质粒并保存。培养HEK-293细胞至80%融合度后,利用Lipofectamine 2000试剂盒 (美国 Invitrogen公司) 转染上述质粒至细胞。培养48 h后利用荧光显微镜 (德国徕卡公司) 观察细胞形态。
2 结果
2.1 临床资料
患者,男,30岁,既往有发作性软瘫7年的病史,常因为天气变凉而诱发,实验室检查发现间断低钾血症 (入院后血钾3.33 mmol/L),长期自行补钾治疗。家族史:父亲有类似表现,但未经系统诊治。患者无肾损伤相关用药史和其他肝脏疾病、自身免疫性疾病病史,完善了其他低钾血症病因的相关检查,如肾上腺增强3D-CT未见异常,促肾上腺皮质激素-皮质醇节律和测定值在正常范围,肾素-血管紧张素-醛固酮水平正常,风湿抗体系列正常。因临床表现不典型,且存在家族史,故行二代测序明确低钾原因。
2.2 基因检测结果
全外显子组测序提示,患者携带一处位于SLC4A1基因 (NM_000342) 第11外显子上的杂合性突变 (c.1162C>T,p.Arg388Cys)。Sanger测序验证了本突变,并且对患者亲属的相应位点进行了测序,发现患者父亲携带相同突变 (图1A、1B)。
2.3 细胞学实验
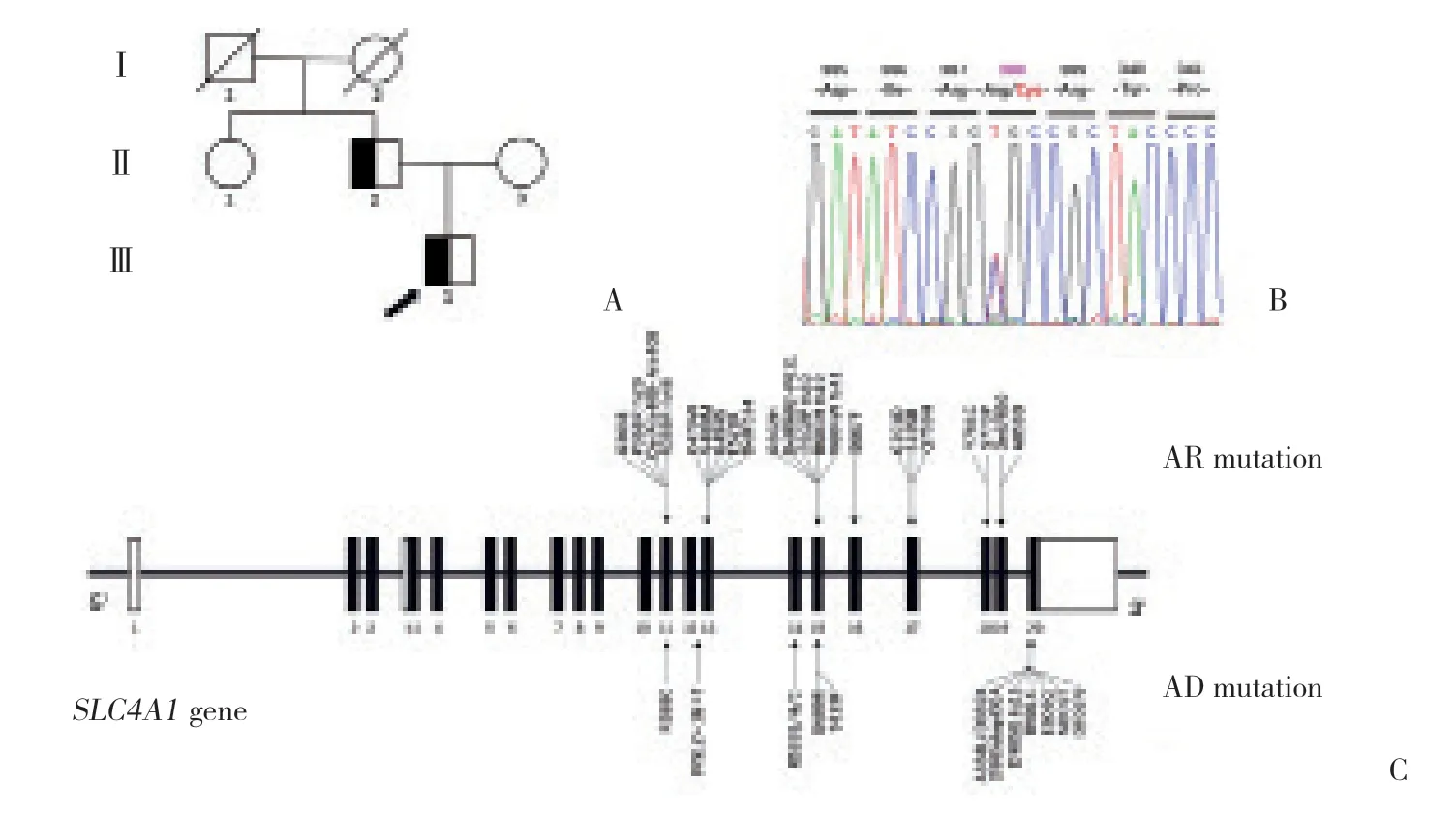
图1 患者的家系图和基因测序结果和SLC4A1基因突变示意图Fig.1 Pedigree of the kindred and sequencing result of SLC4A1 gene with schematic diagram of the mutation sites
将携带野生型和突变型SLC4A1基因序列的GV230质粒转染到HEK-293细胞后,培养48 h后在荧光显微镜下观察发现,转染野生型SLC4A1质粒的细胞可见SLC4A1绿色荧光蛋白分布于细胞表面,提示蛋白合成后可转运至正常位置;而转染突变型SLC4A1质粒的细胞可见SLC4A1绿色荧光蛋白分布在细胞质内,无法达到细胞表面,不能发挥正常作用 (图2)。

图2 转染野生型kAE1和野生型kAE1质粒的HEK293细胞 ×200Fig.2 HEK293 cells transfected with wild-type kAE1 and wildtype kAE1 plasmid ×200
3 讨论
dRTA分为原发性和继发性,继发性因素包括慢性肝脏疾病、自身免疫性疾病、肾移植、药物性肾损伤等,多见于成人。而原发性dRTA多为婴幼儿起病,甚至不能成活,成人起病较为少见,多为不完全性或非经典性,因此较难鉴别,需细致除外继发性因素。典型dRTA的临床表现包括软瘫或抽搐等低钾血症表现、骨骼异常表现 (成人骨软化症、儿童佝偻病、骨折、骨痛等)、感觉神经性耳聋等,实验室检查会发现阴离子间隙正常的代谢性酸中毒、低钾血症、高尿钙、尿液pH值增高,必要时可完善氯化铵试验,判断肾小管的酸化功能。影像学检查会发现肾脏钙质沉着 (常见) 和肾结石 (少见),骨骼呈佝偻病样改变,在部分听力减低的患者中,内耳CT检查可发现前庭导水管扩张[5]。本文的患者以低钾血症就诊,起病年龄较早,并且与其父亲有相似的临床表现,因此考虑遗传性疾病的可能性。由于患者症状不典型,并无其他dRTA临床表现,因此并未首先考虑遗传性dRTA,而是直接进行了二代测序以明确低钾血症的病因,这体现了二代测序在诊断不典型遗传性疾病中的优势。
以往认为遗传性dRTA的致病基因主要为ATP6V1B1基因、ATP6V0A4基因和SLC4A1基因,其中最多见的为ATP6V1B1基因和ATP6V0A4基因 (共占70%),SLC4A1基因突变约占15%左右[2,5],新近有个例报道FOXI1基因[3]和WDR72基因突变也可以导致遗传性dRTA[4],此外还有15%的患者家系存在未知的基因突变位点。由于遗传性dRTA的致病基因较多,因此在临床中可疑遗传性dRTA的患者同样应推荐进行性价比较高的二代测序进行基因诊断。
通常ATP6V1B1、ATP6V0A4、FOXI1和WDR72基因突变导致的dRTA的临床表现 (代谢性酸中毒和低钾血症) 要比SLC4A1基因突变导致的dRTA严重 (前3个基因突变的临床表现常伴随耳聋)[3-4,8]。SLC4A1基因编码的蛋白可在肾脏和红细胞膜中表达,在红细胞膜中表达的AE1又称为带3蛋白,是红细胞膜的重要组成成分,因此部分突变类型可造成遗传性球形红细胞血症,从而导致溶血性贫血。在肾脏的α闰细胞中表达的AE1又称为kAE1,可使碳酸氢根与氯离子交换并重吸收入血液。因此,其功能受损会导致酸中毒、低血钾、尿钙增加等一系列dRTA的临床表现。
目前文献已报道导致dRTA的SLC4A1基因突变有36处,遗传模式根据突变类型不同可表现为常染色体显性遗传 (autosomal dominant,AD) 或常染色体隐性遗传 (autosomal recessive,AR)[9-10],热点区域为第11、13、15和20外显子。AD dRTA的发生机制为kAE1二聚体中的杂合性突变蛋白导致正常kAE1蛋白的运输障碍,又称之为“显性-隐形效应”,如R589H和S613F[11],以及集中于C端第20外显子的突变,如A888L/D889X (del20bp)[7]、R901X (ins13bp)[12]、D902N[13]、D905dupCGA (ins3bp)[14]、D905G fs15(ins1bp)[15]、E906Q[16]和M909T[17]。而导致AR dRTA最常见的位点是位于第17外显子的G701D[14,18]。本例患者的SLC4A1基因突变R388C位于第11外显子,系kAE1蛋白N端的双性螺旋1 区域 (H1区域),属于高度保守区域,推测R388C可能会引起kAE1构像上的变化,造成其与野生型kAE1形成的杂合二聚体停留在内质网内,无法正常转运至胞膜发挥正常的作用,这也在本研究的细胞实验中得到了证实,在遗传模式和致病机制方面都符合AD 模式的dRTA。SLC4A1基因突变导致的dRTA临床表现异质性较强,甚至相同突变的表现也不尽一致,通常AD模式的临床表现要轻于AR模式[1]。本例患者及其父亲起病时间长,就诊时无酸中毒表现,仅表现为间断低钾血症、碱性尿和高尿钙,父亲甚至常年未正式就医,这与其他AD模式的dRTA临床表现相似[6-7,13]。
dRTA的治疗首要是要及时纠正酸中毒和低钾血症,从而避免出现急性症状和降低慢性并发症的严重程度 (如生长迟缓、肾钙质沉着、骨软化、肾小球滤过率下降等),通常选择碱性药物如枸橼酸钾、枸橼酸钠等;其次是要定期随访,监测血气分析、血离子、尿离子、肾脏超声、骨密度、听力等;三是建议做基因检测,可以选择二代测序,并合理的进行遗传咨询[1]。
综上所述,本文对文献中已报道的SLC4A1基因突变进行了归纳总结,并对前期发现的位于SLC4A1基因H1区域的一处突变R388C进行了发病机制的探讨,细胞学实验证实突变的kAE1蛋白无法达到细胞表面发挥正常的离子转运功能,提示本突变是造成AD dRTA的分子基础。
