超临界甲醇中氧化脱除模型物中噻吩硫的研究
2020-08-07许金山许晓斌曹发海
陈 圣, 陈 辉, 许金山, 许晓斌, 许 军, 曹发海
(1. 华东理工大学 大型工业反应器工程教育部工程研究中心, 上海 200237;2. 中国石油化工股份有限公司, 北京 100728; 3. 中石化齐鲁分公司研究院, 山东 淄博 255400)
1 前 言
在原油劣质化和重质化的全球性趋势下[1],延迟焦化工艺所生产的石油焦硫含量居高不下,而这部分硫最终以硫氧化物(SOx)的形式排入大气[2-3],造成污染。2016 年我国新环境法对出厂石油焦硫含量作了更严格的规定。因此,对现有延迟焦化石油焦进行脱硫有现实要求与意义。然而石油焦硫形态复杂,直接脱除非常困难,且脱除成本高,还可能影响石油焦质量[2,4]。渣油是生产石油焦的原料,关于渣油脱硫已有大量的研究[5-8],若能从源头渣油中脱硫不仅可使延迟焦化石油焦硫含量达标,还可降低其他延迟焦化产品的硫含量。
加氢脱硫(HDS)是最传统也是最常用的石油馏分脱硫技术[7-8]。通过在一定压力和温度下进行催化加氢,将硫化物中的S 以H2S 的形式从体系分离。但由于空间位阻效应,具有高稳定性的噻吩硫与催化剂相互作用时活化程度低,难以直接氢解脱硫[9],尤其是4-MDBT、4,6-DMDBT[10],而噻吩硫却是渣油中主要的含硫化合物。
氧化脱硫技术(ODS),在氧化剂作用下选择性地将噻吩硫氧化为极性更强的亚砜或砜类化合物[6,11-12],使硫化物被萃取至极性相从而实现与油品的分离。相较于加氢脱硫,氧化脱硫技术更易脱除噻吩硫,且加氢脱硫难以脱除的甲基取代噻吩硫反而具有更强的氧化反应性[13-15]。另外,氧化脱硫在相对低温、低压的条件即可实现对噻吩类硫化物的转化脱除。但目前ODS 主要还是应用于轻质油品的脱硫,而对于减压渣油等重质油,由于其高黏性、低流动性带来的阻力效应使反应效率降低,鲜有文献涉及渣油氧化脱硫。有些氧化剂为水溶性氧化剂如H2O2[16-18],此类氧化剂不溶于渣油,即便采用搅拌装置也很难保证反应物充分混合与反应。
针对重质原料油流动性差、反应活性低的问题,文献报道了超临界技术在渣油的应用[19-23]。在超临界条件下,介质的黏度可与气体相媲美,从而加速了传质过程,并使一些催化剂长时间保持高活性[24-25]。超临界态下的渣油与溶剂的混合物为均相状态,可解决水溶性氧化剂与渣油接触不充分的问题,使氧化、萃取过程充分进行。此外,超临界态下的反应能够改善渣油的四组分分布,增加轻组分的含量,提升渣油质量[19-22],这对渣油延迟焦化具有积极意义。
综上所述,如能将ODS 技术与超临界脱硫技术有机结合,可实现延迟焦化原料渣油脱硫,从而降低产品石油焦硫含量。YUAN 等[26]的工作指出,将氧气引入超临界脱硫体系可提高渣油的脱硫率。JAVADLI等[5]将ODS 的氧化、萃取步骤合并,缩短了工艺,并发现其具有一定的脱硫效果。这些工作表明超临界氧化脱硫具有可行性。基于上述原因,提出将超临界与氧化脱硫耦合,即将ODS 过程的氧化、萃取过程与超临界状态相结合,使氧化与超临界萃取在同一单元内进行。并且,利用超临界态消除相际传质阻力,增加传递效率,而反应结束后冷却,两液相又自动分离,硫化物被萃取至极性相,从而实现渣油深度脱硫。为了降低研究的复杂性,以二苯并噻吩为模型化合物,考察非临氢无催化条件下,反应温度、反应压力、反应时间及氧化剂量等工艺条件对超临界甲醇中氧化脱硫工艺的影响。此外,研究非临氢环境下超临界甲醇中氧化脱硫催化剂体系、相关催化脱硫反应机理,为重质燃料油超临界氧化脱硫工业化应用提供理论依据。
2 实验部分
2.1 主要实验药品
二苯并噻吩(99%),甲醇(AR),购于上海泰坦科技股份有限公司;正十四烷(99%),购于上海笛柏化学品技术有限公司;无水氯化钴(II) (98%),购于研峰科技(北京)有限公司;辛醛(99%),钨酸钠(98%),Co3O4(II, Ⅲ ) (99.5%),购于上海麦克林生化科技有限公司;过氧化二叔丁基(97%),购于上海阿拉丁生化科技股份有限公司。
2.2 实验仪器
红外光谱仪:美国尼高力公司 6700 型傅里叶红外光谱仪(涂层);气质联用仪:美国安捷伦公司7890A-5975C 型气相色谱/质谱联用仪(样口温度300 ℃;检测器温度:320 ℃;色谱柱温度:280 ℃)。微机库仑测硫仪:江苏国创分析仪器有限公司CLS-3000 型(炉温1 000 ℃)。微型高压反应釜:上海岩征仪器YZPR-100 (100 mL)。
2.3 实验步骤
2.3.1 模型物配制
称取一定量纯度为99% 的二苯并噻吩(DBT),用正十四烷溶解并在200 mL 容量瓶中定容。配制硫质量含量为0.5% 的模型油,然后用微机库伦测硫仪标定其浓度。
2.3.2 超临界氧化
模型油初始硫质量浓度为0.5%,模型油用量15.0 mL,超临界介质甲醇用量15.0 mL,反应前排除体系内空气,并用 N2作保护气。反应结束后,先用碱液吸收釜内气体,后将反应产物液-液两相分离,分别称取质量并用微机库仑测硫仪测定硫含量,计算总硫平衡;同时取少量产品去红外分析与气相色谱/质谱联用分析。
3 结果与讨论
3.1 无催化超临界氧化脱硫
对于超临界氧化脱硫过程,反应温度、反应压力、反应时间、氧化剂及用量是影响脱硫率的重要因素。在临界温度与临界压力附近,略微改变温度或压力可能给整个体系带来较大影响。因此本文先在不使用催化剂的情况下,对这些因素进行考察,并选择最优的反应条件进行后续研究。
3.1.1 反应温度对脱硫率的影响
反应温度对脱硫率的影响如图1 所示。其反应条件为:反应压力8.0 MPa,反应时间3 h;所用氧化剂为辛醛,氧/硫(O/S)摩尔比为3:1。甲醇的临界条件为:240 ℃,7.95 MPa。从图1 可知,在甲醇临界点以下的 200~240 ℃,脱硫率随温度变化不明显。在甲醇临界点附近有一段脱硫率快速增长区域,即240~260 ℃,这是由于超临界态下两相均一化,体系的传质速率增加,反应物碰撞概率增加,因此反应更充分,脱硫速率快速增加;在超临界状态下,温度越高,形成的中间活性物越多,中间活性物质与噻吩硫化物的碰撞概率增加,噻吩硫化物被氧化速度加快,噻吩硫化物的脱除率随之增大。240~260 ℃ 脱硫率上升速度远大于260~280 和280~300 ℃。但温度继续升高,蒸汽压力增大,增加反应危险性;并且由于温度越高能耗越高,体系也更难控制,故温度只考察到300 ℃。反应温度为260 ℃ 脱硫效率最高,并且工业上减压渣油出塔温度也更接近该温度,因此将其作为后续脱硫温度条件。
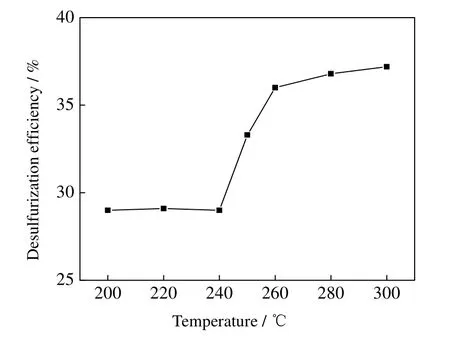
图1 脱硫率随反应温度变化Fig.1 Effects of reaction temperature on DBT removal
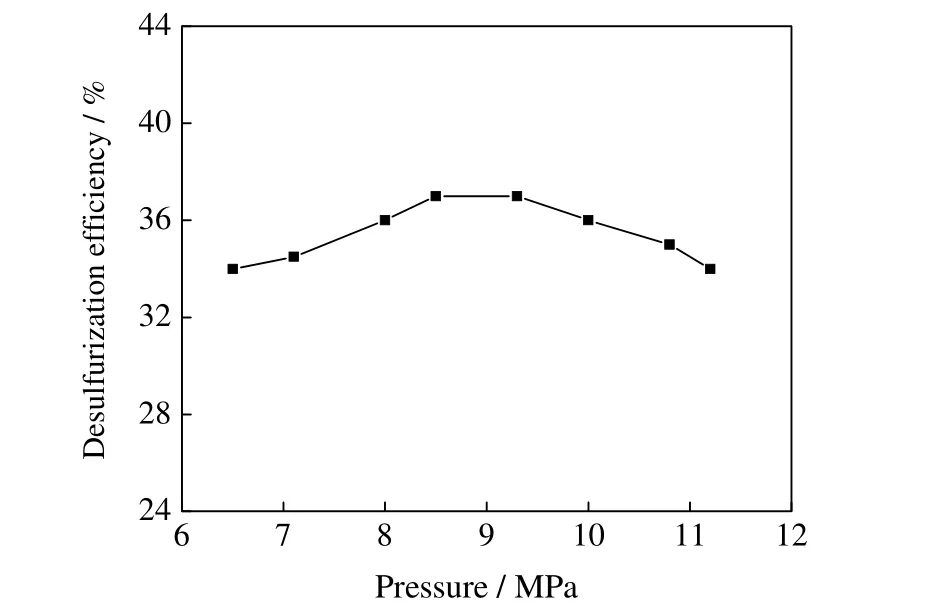
图2 脱硫率随反应压力变化Fig.2 Effects of reaction pressure on DBT removal
3.1.2 反应压力对脱硫率的影响
反应压力对脱硫率的影响如图2 所示。其反应条件为:反应温度260 ℃,反应时间3 h;氧化剂为辛醛,O/S 为3:1。从图2 可知,脱硫率总体随反应压力变化不大。在甲醇临界点附近,脱硫率先随反应压力增大而增大;反应压力增至8.5 MPa 时,脱硫率达到37%,继续增加压力到9.3 MPa 以上,脱硫率逐渐下降。临界点效应相较于反应温度不明显,在临界点上方压力过高反而不利于反应脱硫。在后续的考察中,反应压力控制在8.5~9.0 MPa。
3.1.3 反应时间对脱硫率的影响
反应时间对脱硫率的影响如图3 所示。其反应条件为:反应温度260 ℃,反应压力9.0 MPa;所用氧化剂为辛醛,O/S 为3:1。由图3 可知,脱硫率随反应时间先增加,后趋于稳定。随着反应进行,反应快速进行,反应进行到3 h 时,脱硫率达到峰值,此时噻吩硫化物的脱除率接近40%。继续进行反应,脱硫率保持稳定。因此将3 h 作为后续研究中的脱硫时间。
3.1.4 氧化剂对脱硫率的影响
氧化剂是氧化脱硫的重要影响因素。考察氧化剂使用量对脱硫率的影响,不同氧化剂的脱硫效果。醛类是一种弱氧化剂,有时也作还原剂。MURATA 等[27-28]的工作提到醛类作为一种牺牲剂参与氧化脱硫反应,不使用或减少醛类用量会导致反应无法进行或反应速率下降。在本研究中,为避免强氧化剂带来的副反应,甚至影响油品质量,首先考察醛类作氧化剂的脱硫效果。
氧化剂用量对脱硫率的影响如图4 所示。其反应条件为:反应温度260 ℃,反应压力9.0 MPa,反应时间3 h。由图4 可知,这两种醛作氧化剂时,随着氧硫摩尔比O/S 的升高,脱硫率先增加,后趋于稳定。两种氧化剂的脱硫率都在O/S 为3 时达到峰值。相较于辛醛,用肉桂醛作氧化剂时,在O/S 达到3 之前,随着O/S 的增加其脱硫率增长先快后慢,在O/S 为3~5,二者下降趋势基本一致。辛醛属于脂肪醛,肉桂醛属于芳香醛,由于芳香醛还原性弱于脂肪醛,其氧化性应强于脂肪醛,在图中表现为前期的快增长速率。当O/S 达到3 时,两种氧化剂的脱硫率开始稳定;O/S 继续增大,脱硫率无明显变化。因此将O/S = 3 作为后续脱硫氧硫摩尔比。
文献报道的氧化脱硫氧化剂种类繁多,如过氧化物类的过氧化氢(30%),广泛用于氧化脱硫研究中[18,29-30],其他过氧化物类氧化剂如过氧化环己酮[31],过氧化羟基异丙苯等[32-33],高价态金属盐类如高铁酸盐[34-35],以及特殊氧化剂如硅胶[36]。许多文献也提到了用臭氧[37],氧气[18,38-39]或空气[5,15]作氧化剂,但由于本研究在较高温度压力下进行,引入气体涉及爆炸极限的问题,在工业上应用十分危险,因此在本研究中不考虑该类氧化剂,并选择上述部分氧化剂作考察。
不同氧化剂对脱硫率的影响如图5 所示。其反应条件为:反应温度260 ℃,反应压力9.0 MPa,反应时间3 h,O/S 为3:1。由图5 可知,在无催化剂作用下,过氧化二叔丁基 DTBP 的脱硫率最高,达 42%。过氧化氢(30%),过氧化苯甲酰以及过氧化羟基异丙苯等过氧化物类氧化剂在该反应条件下脱硫率并不理想,这是由于在高温下这些氧化剂容易分解,其中过氧化氢(30%)作氧化剂脱硫率不到 30%。而造成这近 30% 硫脱除率的原因,主要是过氧化氢等氧化剂分解前产生的部分氧化效果,分解产物可能具有一定氧化作用。过氧化二叔丁基亦为过氧化物类氧化剂,但其相对其他过氧化物不易分解,因此脱硫率较高。

图3 脱硫率随反应时间变化Fig.3 Effects of reaction time on DBT removal

图4 脱硫率随O/S 摩尔比变化Fig.4 Effects of O/S molar ratio on DBT removal
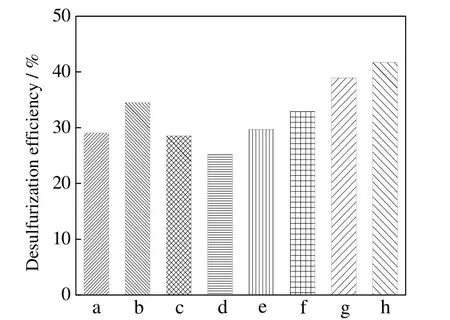
图5 不同氧化剂脱硫率对比Fig.5 Effects of oxidant type on DBT removal
3.2 超临界甲醇中催化氧化脱硫
催化氧化体系在氧化脱硫反应中发挥着重要作用。关于催化氧化脱硫的催化剂研究已有非常多的文献报道。其中金属类催化剂[38-39],如Co、Mn;金属氧化物[15,39]如Co3O4、MnO2等。一些研究提到了氧化剂、催化剂与牺牲剂结合使用,比如用醛类作牺牲剂[28,39-40]。其他催化剂[14,41-42],如多金属含氧酸,离子液体,碳纳米管脱硫效果也十分显著。在这些催化剂体系中,有些不使用载体,有些则用传统载体如二氧化硅和氧化铝负载催化剂,但这些载体在超临界反应环境中严重降解[43]。本文结合上述文献及实际,考察Co3O4,MnO2,Na2WO4,CoCl2等4 种催化剂,并比较复合氧化剂与单一氧化剂的脱硫效果。
3.2.1 催化剂及用量对脱硫率的影响
催化剂用量对脱硫率的影响如图6 所示。其反应条件为:反应温度260 ℃,反应压力9.0 MPa,反应时间3 h,氧化剂为辛醛,O/S 为3。
从图6 可知,氯化钴的催化效果优于钨酸钠,钨酸钠对脱硫率几乎无影响。随着催化剂与硫化物的摩尔比(C/S)增加,脱硫率增加到一定程度后趋于稳定,当C/S 为1:4 时,脱硫效果最佳;但实际上当C/S达到1:10 后,继续增大催化剂用量带来的脱硫率升高并不明显,却要消耗双倍甚至更多的催化剂量。为达到最高的催化剂使用效率,在后续的研究中将使用C/S 为1:10 的催化剂用量。

图6 脱硫率随C/S 变化Fig.6 Effects of C/S molar ratio on DBT removal
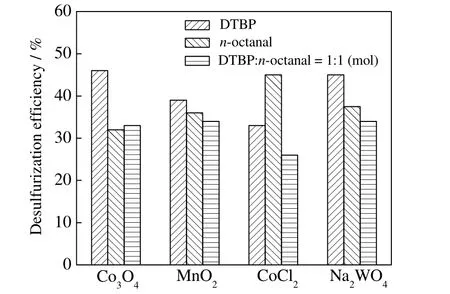
图7 不同催化氧化体系脱硫效果对比Fig.7 Effects of catalytic oxidation systems on DBT removal
3.2.2 催化氧化体系对脱硫率的影响
不同催化氧化体系对脱硫率的影响如图7 所示。其反应条件为,反应温度260 ℃,反应压力9.0 MPa,反应时间3 h,O/S 为3,C/S 为1:10。
由图7 可知,当使用两种氧化剂或将其中的醛类视作牺牲剂时,其脱硫效果不如单独使用其中某个氧化剂。过氧化二叔丁基/Co3O4、辛醛/CoCl2、过氧化二叔丁基/Na2WO43 种催化氧化体系脱硫效果较好,脱硫率分别达到了47%,45%,45%。MnO2作催化剂时,脱硫效果随氧化剂类型变化不大。
与文献报道中模型油的氧化脱硫率相比,本文整体脱硫率相对较低,其可能原因如下:多数文献报道中模型油的氧化脱硫率可达 80%,甚至更高,但其模型油的硫含量很低,仅为百万分之一级别[14-15,27,29,34,36,44],而本文所用模型油含硫质量达0.5%,两者初始硫含量不在同一个数量级。初始硫含量较高的油品,其脱硫率则相对较低。OTSUKI 等[45]对初始含硫质量为1.35% 的直馏轻瓦斯油(SR-LGO)氧化脱硫,脱硫率可达52%;而初始硫含质量为2.17% 的减压瓦斯油(VGO),脱硫率仅为32%。TIMKO 等[17]对初始硫质量含量分别为720×10-6和1 400×10-6的3773 和4177 燃料脱硫,前者脱硫率可达94% 以上,后者脱硫率仅为75%。初始硫水平,一定程度上影响了脱硫率。JAVADLI[5]对初始硫含量为5% 加拿大冷湖沥青氧化脱硫,最佳脱硫率也仅为47%。另外,苛刻的超临界条件也可能造成脱硫率偏低,氧化剂在高温、高压环境下氧化性能可能会受到破坏,导致脱硫率在一定程度上下降。
3.3 硫转移机制
关于氧化脱硫的机理,研究者们给出了不同的见解,氧化产物有SO2[5], SO24-[46],亚砜,砜类等。多数文献认为噻吩硫氧化产物为亚砜或砜类[14-15,39,44]。本文对反应产物进行分析,探究催化氧化反应机理。
测得吸收瓶中碱液硫含量为0,表明反应后气相中无硫化物,即反应后硫分布在液-液两相产物中;根据气-液-液硫守恒关系检验上述结果,证实反应后硫化物的确分布在液-液两相中。采用红外光谱对反应前后的甲醇进行分析。图8 为反应前后甲醇的红外吸收光谱图,其中反应后1 为辛醛-氯化钴体系氧化后的甲醇相产品,反应后2 是过氧化二叔丁基/Co3O4体系氧化后的甲醇相产品。
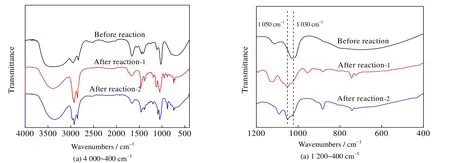
图8 反应前后甲醇相红外吸收光谱对比图Fig.8 Comparison of FT-IR spectra of methanol before and after reactions
由图8 可知,1 030 cm-1处峰在反应后发生偏移,在1 050 cm-1处产生了一个新峰。而亚砜在红外光谱1 030~1 070 cm-1处有特征吸收峰,在ZHANG 等[16]的工作中提到了二苯并噻吩对应亚砜(DBTO)的红外吸收光谱在1 047.16 cm-1,因此上述新峰可能为二苯并噻吩的氧化物,即二苯并噻吩5-氧化物(DBTO)。其可能的反应机理如图9 所示。
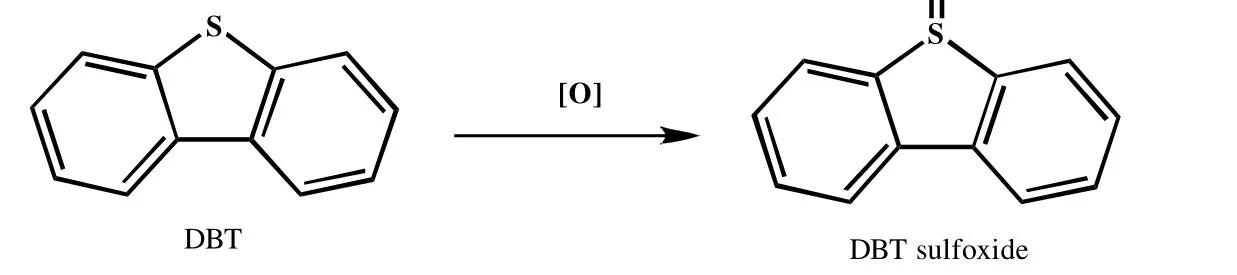
图9 二苯并噻吩氧化反应机理简图Fig.9 Mechanism of DBT oxidation reaction
为进一步确定反应产物甲醇相中的硫形态,本文对过氧化二叔丁基/Co3O4体系氧化后的甲醇产品进行了气相色谱分析。图10 为反应后产物中甲醇的气相色谱测试谱图。
从图10 可知,反应产物甲醇相中含有DBTO 的峰,因此可以得出二苯并噻吩氧化后为 DBTO。由于生成的亚砜极性增大,由相似相溶原理可知,亚砜更易溶于极性介质甲醇中。超临界甲醇中氧化脱硫结合了氧化脱硫这一脱硫机制与超临界态的相态均一性,实现了硫化物的脱除。
超临界甲醇中氧化脱硫机制如图11 所示。其主要由4 个状态。a:反应前。此时两相处于分离状态,硫化物二苯并噻吩存在于模型油中。b:亚临界态。在反应釜升温过程中,部分二苯并噻吩与氧化剂反应,生成的亚砜向极性溶剂甲醇偏离。c:超临界态。在超临界态下,两相趋于均一化,氧化和萃取过程有效、快速进行;d:反应结束降温。该状态下两相分离,亚砜溶于极性更大的甲醇相中,从而实现硫化物与油品分离。
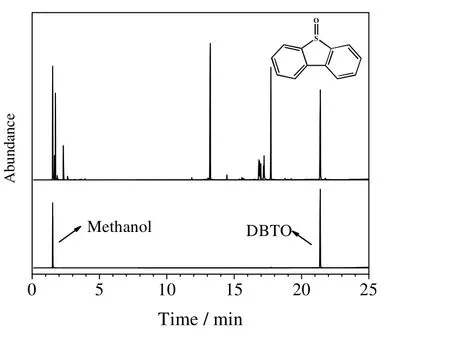
图10 产物甲醇与标准物气相色谱图对比Fig.10 Comparison of GC spectra of standard DBTO (MeOH) and reaction products
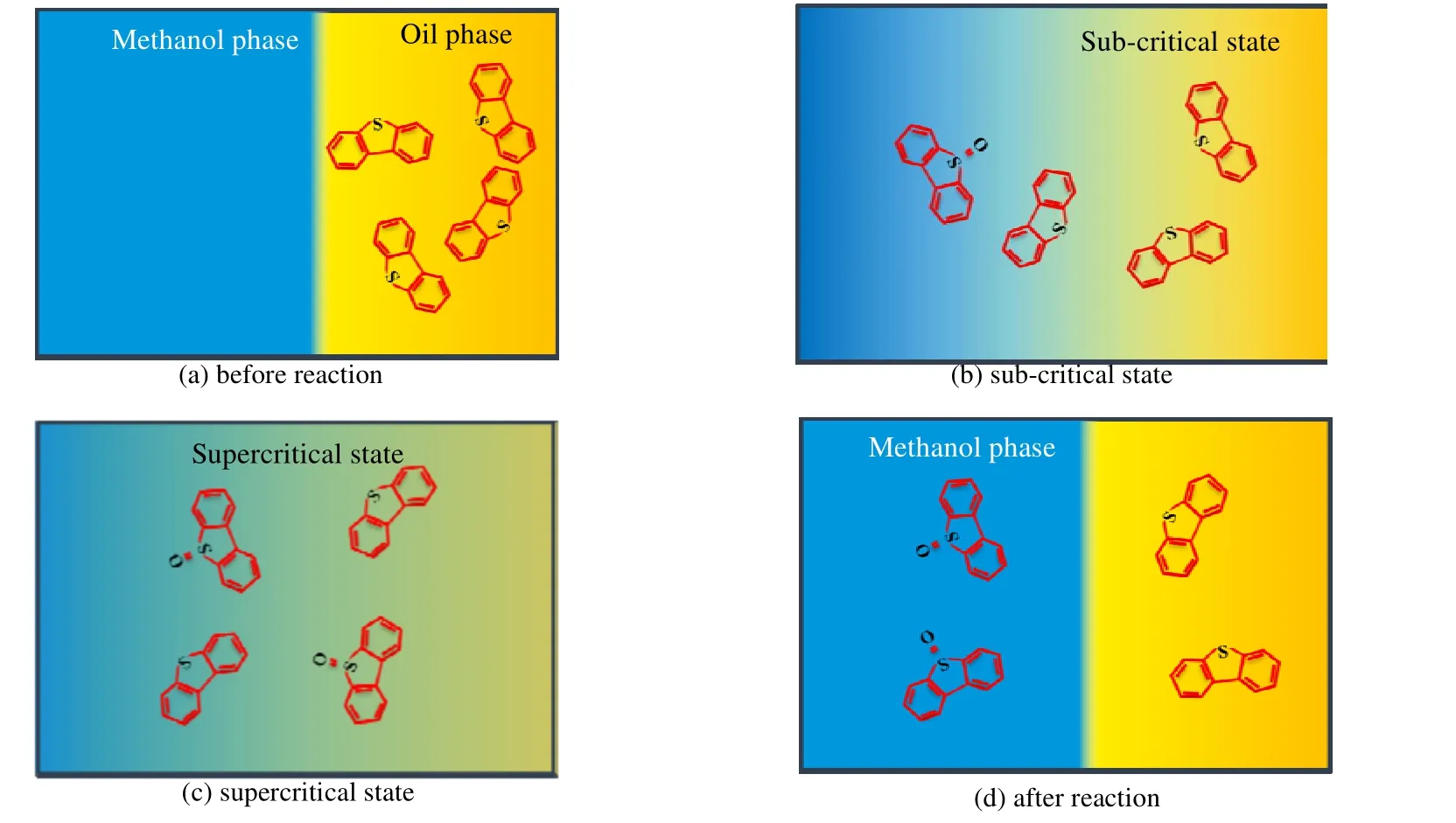
图11 超临界甲醇中氧化脱硫机制Fig.11 Oxidative desulfurization process in supercritical methanol
4 结 论
以二苯并噻吩作为模型物探究了超临界甲醇中氧化反应脱硫机理。不使用催化剂时,最优反应条件为:反应温度260 ℃,反应压力8.5~ 9.0 MPa,反应时间3 h,O/S 摩尔比为3:1,过氧化二叔丁基作氧化剂在此条件下脱硫率可达42%,在甲醇临界点附近,温度对脱硫影响大,超临界效果显著;采用催化氧化体系时,C/S 摩尔比为1:10 时催化剂效率最高,过氧化二叔丁基/Co3O4、辛醛/CoCl2、过氧化二叔丁基/Na2WO4催化氧化体系的脱硫率较高,分别达47%,45%,45%。FT-IR 与GC-MS 结果表明过氧化二叔丁基/Co3O4催化氧化二苯并噻吩的氧化产物为对应亚砜,即二苯并噻吩5-氧化物。
