鞍区朗格汉斯细胞组织细胞增生症临床特点分析
2020-08-07王志成朱建宇张毅李晓旭刘杰朱惠娟姚勇潘慧邓侃
王志成 朱建宇 张毅 李晓旭 刘杰 朱惠娟 姚勇 潘慧 邓侃
朗格汉斯细胞组织细胞增生症(LCH)是一种病因不明的组织细胞浸润性疾病,因肿瘤细胞在组织学形态和免疫表型与皮肤和黏膜朗格汉斯细胞相似而得名,但是从基因表达阵列数据看,病态朗格汉斯细胞起源于髓样树突状细胞,并非由皮肤和黏膜朗格汉斯细胞转化而来[1]。LCH好发于儿童和青少年,可以累及单器官或多器官,依次为骨骼(80%)、皮肤(33%)、垂体(25%)、肝脏(15%)、脾(15%)、造血系统(15%)、肺(15%)、淋巴结(5%~10%)和中枢神经系统除垂体外的其他部位(2%~4%)[2]。受累部位不同,临床症状略有差异,可以表现为发热、乏力、消瘦、骨痛、皮损、中枢性尿崩症、黄疸、咳嗽、胸痛等,明确诊断依靠组织病理学检查。累及鞍区的LCH 临床罕见,主要表现为中枢性尿崩症、头痛、视力视野损害、腺垂体功能减退症、下丘脑相关症状等,其中中枢性尿崩症为最常见症状。对于以中枢性尿崩症发病、头部MRI 提示鞍区占位性病变、拟诊LCH 的患者,考虑到脑组织活检术的风险相对较高,积极寻找可能支持诊断的颅外病变即显得十分重要。单纯累及鞍区者十分罕见,在以中枢性尿崩症发病的LCH 中仅占9%[3],并且既往多为个案报道。加之缺少特异性临床表现和可用于组织活检的颅外病变,大多数患者仅能通过定期随访,直至鞍区病变进展而不得不接受组织活检、或出现颅外病变、或接受经验性治疗并经MRI监测病情变化。本文拟对中国医学科学院北京协和医院神经外科近年收治的8例累及鞍区的LCH病例的临床特点进行总结,并探讨鞍区组织活检术的安全性。
临床资料
通过中国医学科学院北京协和医院信息系统检索获得2011 年11 月至2019 年11 月在神经外科住院并符合以下纳入标准的病例共计8 例:头部MRI 提示鞍区占位性病变;于神经内镜下行经鼻蝶入路(ETA)或扩大经鼻蝶入路(EETA)鞍区病变组织活检术证实为LCH。男性4 例,女性4 例;年龄为10 ~ 47 岁,中位年龄15.50(12.25,38.00)岁,其中 <18 岁者 5 例、≥ 18 岁 3 例;发病至确诊时间 2 ~ 50 个月,中位时间4.00(2.25,28.75)个月(表1)。(1)症状与体征:主要以多饮、多尿、烦渴、夜尿增多等中枢性尿崩症表现发病;其中,7 例同时伴有食欲减退、乏力、性欲减退、月经紊乱、闭经、遗精减少等腺垂体功能减退症状,1 例伴头痛,1 例表现为下丘脑相关症状如间断性发热(37 ~38 ℃),多出现在早餐或午餐后,约至黄昏时(18∶00)自行退热(表1)。(2)实验室检查:所有患者血清和脑脊液甲胎蛋白(AFP)或β-人绒毛膜促性腺激素(β-hCG)水平均于正常参考值范围(表2)。(3)垂体MRI 表现:占位性病变累及范围包括鞍内及垂体柄(例2、例6、例8),垂体柄及下丘脑(例5、例7),鞍内、垂体柄及下丘脑(例3),鞍内、左侧颞叶内侧和胼胝体(例4),以及单纯垂体柄受累(例1)。T1WI 神经垂体高信号消失,呈等信号者8 例;T2WI 呈等信号者6 例(例1 ~ 例4、例6、例8)、低信号1例(例5)或混杂信号1例(例7);增强扫描呈均匀强化者6 例(例1、例3 ~例7),不均匀强化者2 例(例2、例8)。8 例患者均未发现颅外病变(表2)。(4)组织活检术:对于MRI 显示占位性病变累及鞍内的患者(例2 ~例4、例6、例8)于神经内镜下经鼻蝶入路行鞍内病变组织活检术,术前30 min 和术后72 h 常规应用头孢呋辛钠1.50 g/次(2 次/d)静脉滴注预防感染;病变完全位于鞍上者(例1、例5、例7)经神经内镜下扩大经鼻蝶入路行鞍上病变组织活检术和脂肪浴缸塞联合鼻中隔黏膜瓣鞍底重建术,术前30 min 和术后72 h 常规予头孢他啶2 g/次(2 次/d)静脉滴注预防感染。(5)术后并发症:本组仅1例(例5)患者组织活检术后1 d出现双眼颞侧视物成双、模糊,术后3 d 上述症状缓解,术后6 d 上述症状完全消失,考虑为短暂性下丘脑反应。无一例患者术后1 个月内发生脑脊液鼻漏、中枢神经系统感染、非计划二次手术、死亡等其他手术相关并发症(表2)。

表1 8例鞍区朗格汉斯细胞组织细胞增生症患者社会人口学资料和临床表现Table 1. Demographic data and clinical manifestations of 8 patients with sellar LCH

表2 8例鞍区朗格汉斯细胞组织细胞增生症患者实验室、MRI、组织活检术和术后并发症Table 2. Laboratory, MRI, biopsy and postoperative complications of 8 patients with sellar LCH
典型病例
患者(例8) 女性,16 岁。因多饮、多尿4 个月,月经不规律 2 个月,于 2019 年 4 月 3 日入院。患者入院前4 个月(2018 年12 月)无明显诱因出现多饮、多尿,每日饮水量约10 L,白天小便1 次/h、夜尿3 ~4次;入院前2 个月开始出现月经不规律,偶有恶心,但无头痛、呕吐、发热、乏力,以及体毛脱落、性欲改变和异常泌乳等改变,自觉无视力下降、视野缺损等症状,外院垂体MRI 检查显示垂体形态饱满,信号均匀,冠状位最高径约7.50 mm,垂体柄欠清晰,略增粗,视交叉无明显受压,为求进一步治疗至我院就诊。入院后体格检查未见明显异常。垂体MRI 显示垂体饱满,上缘略膨隆,横径12.70 mm、上下径7.40 mm、前后径8.90 mm,信号均匀,呈等T1、等T2信号,增强后病灶呈明显不均匀强化;垂体柄增粗,横径5 mm、前后径5 mm,视交叉未见受压、移位,神经垂体信号消失,松果体区未见异常信号(图1)。临床诊断为垂体柄占位性病变,原因待查。于2019年4月8日经神经内镜下经鼻蝶入路行鞍内病变组织活检术,术中于鞍内神经垂体部位可见灰白色病变组织,质地较韧,边界欠清晰,血运不丰富,向鞍上生长。组织标本镜下观察,垂体局部纤维组织增生,可见组织细胞、淋巴细胞及嗜酸性粒细胞浸润(图2a);免疫组化染色,上皮细胞表达广谱细胞角蛋白(AE1/AE3);肿瘤细胞表达CD163 和CD1a(图2b),局灶性表达Langerin 和S-100 蛋白(S-100),部分表达CD20 和CD3,散在表达CD68,不表达人婆罗双树样基因-4 蛋白(SALL4),Ki-67 抗原标记指数约为10%;最终病理诊断为LCH。患者术后3 天出院,无手术相关并发症,出院后在外院接受药物化疗(具体方案不详),术后3 个月随访时仍在接受药物化疗。
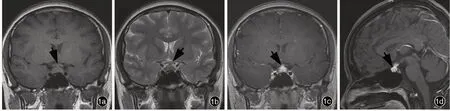
图1 例8 患者术前垂体MRI 检查所见 1a 冠状位T1WI 显示垂体饱满,上缘略膨隆,横径12.70 mm、上下径7.40 mm,呈等信号(箭头所示);垂体柄增粗,横径5 mm;视交叉未见受压移位 1b 冠状位T2WI 扫描垂体呈等信号(箭头所示) 1c 冠状位增强T1WI 扫描,垂体呈不均匀强化、垂体柄明显强化(箭头所示) 1d 矢状位增强T1WI 显示垂体前后径8.90 mm、垂体柄前后径5 mm(箭头所示),神经垂体信号消失,松果体区未见异常信号Figure 1 Head MRI findings before operation of Case 8 Coronal T1WI showed the pituitary gland was plump, the upper edge was slightly bulged, the transverse diameter was 12.70 mm, and the height was 7.40 mm, with isointense signal (arrow indicates). The pituitary stalk was thickened, the transverse diameter was 5 mm, and the optic chiasm was not compressed (Panel 1a). Coronal T2WI showed the pituitary with isointense signal (arrow indicates, Panel 1b). Coronal enhanced T1WI showed that the enhancement of the pituitary was not even, and the pituitary stalk was obviously enhanced (arrow indicates, Panel 1c). Sagittal enhanced T1WI showed the anterior-posterior diameter of pituitary was 8.90 mm, the anterior-posterior diameter of pituitary stalk was 5 mm (arrow indicates), the high signal at the posterior lobe of pituitary was not shown, and the abnormal signal at pineal region was not found (Panel 1d).

图2 例8 患者光学显微镜观察所见 2a 垂体局部纤维组织增生,可见组织细胞、淋巴细胞及嗜酸性粒细胞浸润 HE 染色 ×100 2b 肿瘤细胞表达CD1a 免疫组化染色(EnVison二步法) ×200Figure 2 Optic microscopy findings of Case 8 Local fibrous tissue hyperplasia and infiltration of tissue cells were seen, and lymphocytes and eosinophils were in pituitary (Panel 2a). HE staining × 100 CD1a was positive in tumor cells (Panel 2b).Immunohistochemical staining (EnVison) ×200
讨 论
朗格汉斯细胞组织细胞增生症在儿童和青少年(< 15 岁)人群中的发病率约为4.6/100 万人年[4],成人发病率为 1 ~ 2/100 万人年[5],老年人群偶见个案报道[6],考虑到部分局灶性LCH 具有自发缓解特点,其真实年发病率可能高于文献报道的数据[7]。儿童和青少年(<15 岁)LCH 的确诊中位年龄约为3.5 岁,高峰发病年龄 < 1 岁[4]。本组 8 例鞍区 LCH患者的中位年龄为15.5 岁,其中最小患儿为10 岁,可能与LCH 在年龄更小的患者中发生皮损的概率更高有关[8]。
根据文献报道,约有6%的LCH 患者确诊时中枢神经系统已受累[9],尤以中枢性尿崩症最为常见,而约有15%单纯表现为中枢性尿崩症的患者最终被确诊为 LCH[10]。Prosch 等[3]认为,约有 10.23%(127/1242)的LCH 患者存在中枢性尿崩症,其中42.52%(54/127)确诊时即已存在中枢性尿崩症;在此部分患者中90.74%(49/54)是经颅外病变组织活检术被确诊,其中18.37%(9/49)确诊时即已存在颅外病变。本组8 例患者均以多饮、多尿等中枢性尿崩症发病,男女比例为1∶1,确诊前均未发现其他颅外病变,且无一例存在视力下降、视野缺损或眼球运动障碍等症状与体征,但有7 例表现有食欲减退、乏力、性欲减退、月经紊乱、闭经、遗精减少等腺垂体功能减退症状;病变累及下丘脑区域时,患者可出现嗜食、体温调节紊乱、嗜睡和短期记忆障碍等下丘脑相关症状[11],但本组病变累及下丘脑的3例患者中仅1例出现间断性发热性下丘脑反应。
对于存在中枢性尿崩症的患者,头部MRI 是首选影像学检查方法,T1WI 显示的神经垂体高信号消失被认为是LCH 鞍区受累的典型表现,主要与神经垂体含抗利尿激素的分泌颗粒耗竭有关,本组患者均未见明显的神经垂体高信号征象。然而,神经垂体高信号消失并非LCH 的特异性表现[12]。从病变部位看,本组垂体柄受累者7 例、鞍内受累者5 例,其中鞍内和垂体柄同时受累(3 例)较为常见,其次为垂体柄和下丘脑(2 例)受累;从病变信号看,本组共有4 例(例1、例 3、例4、例 6)呈现T1WI 等信号、T2WI等信号和均匀强化,1 例(例5)呈T1WI等信号、T2WI低信号和均匀强化,1 例(例7)呈T1WI等信号、T2WI 混杂信号和均匀强化,2 例(例 2、例 8)呈现T1WI 等信号、T2WI 等信号和不均匀强化。遗憾的是,尽管MRI 在鞍区占位性病变的临床诊断中具有不可替代的作用,可以反映病变部位、累及范围,并监测疾病进展,但其表现并不具有特异性[13-14]。治疗方面,需根据病变累及范围和病情严重程度选择治疗方法,主要包括局部放射治疗、全身化疗和糖皮质激素治疗[11,15]。
朗格汉斯细胞组织细胞增生症应注意与垂体腺瘤、生殖细胞肿瘤、淋巴细胞性垂体炎等鞍区占位性病变相鉴别。(1)垂体腺瘤:为临床十分常见的颅内肿瘤,约占颅内肿瘤的15%[16],发病率约为0.1%[17]。垂体腺瘤可由腺垂体中任意一种细胞发展而来,分为功能性垂体腺瘤和无功能性垂体腺瘤,功能性垂体腺瘤主要包括垂体生长激素腺瘤、泌乳素腺瘤、促肾上腺皮质激素腺瘤、促甲状腺激素腺瘤、促性腺激素腺瘤等[18],进而导致巨人症和(或)肢端肥大症、停经和(或)泌乳、Cushing病、继发性甲状腺功能亢进症等,从而表现出相应临床症状。实验室检查相关激素水平升高,有助于鉴别诊断。据肿瘤大小可分为垂体微腺瘤(直径<10 mm)和大腺瘤(直径≥10 mm)。无功能性垂体腺瘤由于缺乏特异性激素相关症状,多以局部占位效应而就诊,确诊时大多已进展为大腺瘤,主要表现为头痛、视力下降、视野缺损,以及因压迫导致的腺垂体功能减退症状。由于肿瘤生长较为缓慢,较少导致中枢性尿崩症。垂体腺瘤T1WI呈等或低信号,T2WI呈等信号,强化程度低于正常垂体[19]。治疗方法以外科手术切除或药物治疗为主。(2)颅内生殖细胞肿瘤:较为少见,在中枢神经系统肿瘤中所占比例不足5%、儿童脑肿瘤中占3%~11%,发病率约为0.1/10 万[20-21],男女比例为4 ~ 5∶1,发病高峰年龄为 10~ 20 岁[22],与 LCH 相似。颅内生殖细胞肿瘤好发于松果体和鞍上区[23],位于松果体区的生殖细胞肿瘤通常表现为堵塞脑脊液通道导致的颅内高压症状,位于鞍上区的生殖细胞肿瘤则主要表现为中枢性尿崩症、头痛、腺垂体功能减退症、生长迟缓、视力障碍和视野缺损等。根据组织学形态分为生殖细胞瘤和非生殖细胞瘤性生殖细胞肿瘤(NGGCTs),后者血清和(或)脑脊液AFP 或β-hCG 水平升高,主要累及松果体区[24]。T1WI 呈等或低信号,T2WI 呈等或高信号,增强后病灶呈明显强化征象。由于部分生殖细胞肿瘤临床表现缺乏特异性,其诊断可被延误数月甚至数年[25-26],组织活检术为诊断“金标准”[27]。治疗方法主要包括放射治疗、药物化疗和手术治疗[28-30]。(3)淋巴细胞性垂体炎:是临床较为罕见的自身免疫性疾病,以大量淋巴细胞弥漫性浸润和纤维化为组织学特征[31];发病率约为0.1/100万,好发于女性,特别是孕后期和产后早期女性,中位发病年龄为37 岁[32]。临床表现缺乏特异性,主要表现为中枢性尿崩症、腺垂体功能减退症状、头痛等,尤以中枢性尿崩症常见,发生率约为72%[33]。约有96%患者MRI 表现为垂体柄增粗、78%患者可鞍内和鞍上同时受累。临床诊断较为困难,难以与单纯累及鞍区的LCH 和生殖细胞肿瘤相鉴别,明确诊断依靠组织活检术。治疗方法以糖皮质激素和免疫抑制治疗为主。
鞍区病变的治疗方案可截然不同,因此明确诊断尤为重要。组织活检术仍是诊断LCH 的“金标准”。考虑到颅内组织活检术的相对高风险,积极寻找颅外病变十分重要,但若缺乏支持LCH 诊断的颅外病变时,是否应积极进行颅内组织活检术,以及何时进行活检术即成为诊断与治疗的关键。Prosch 等[34]认为,尽管同时存在诸多看似典型的临床表现,也不应在进行组织病理学检查之前进行临床诊断并治疗。本组8例患者采用神经内镜下经鼻蝶入路或扩大经鼻蝶入路鞍区病变组织活检术,术后仅1 例出现短暂性下丘脑反应,无一例并发脑脊液鼻漏、中枢神经系统感染、非计划二次手术、死亡等不良事件。与开颅手术相比,神经内镜下经鼻蝶入路组织活检术具有美观、对周围脑组织牵拉小、视野开阔和手术损伤小等优势[35-36],因此神经内镜下经鼻蝶入路鞍区占位性病变组织活检术是一种相对安全的方法。我们的临床经验是:(1)对于存在中枢性尿崩症或MRI 提示鞍区占位性病变,拟诊LCH 的患者,应首先完善临床病史、体格检查、血清和脑脊液肿瘤标志物、全身骨显像、头部和胸部X线、腹部超声等检查,积极寻找可能支持LCH 诊断的颅外病变。(2)当缺少颅外病变且鞍区病变直径<5 mm、无视神经压迫症状时,应密切随访,定期复查MRI,一旦内分泌或影像学检查提示病情进展,应尽早行鞍区病变组织活检术。(3)对于缺少颅外病变且鞍区病变直径≥5 mm 或存在视神经压迫症状的患者,除非活检术风险极高,否则均应尽早施行病变组织活检术以明确诊断。
鞍区病变病因多样,部分疾病缺乏特异性临床表现,难以通过无创性检查与单纯累及鞍区的LCH相鉴别,组织活检术仍是诊断的“金标准”。神经内镜下经鼻蝶入路和扩大经鼻蝶入路鞍区病变组织活检术相对安全、有效,可以为临床制定最佳治疗方案和判断预后提供依据。
利益冲突无
