新生儿神经肌肉疾病25例病例系列报告
2020-08-06吴冰冰李西华程国强
张 鹏 杨 琳 赵 蕾 吴冰冰 李西华 程国强
神经肌肉疾病(NMDs)是一组严重致畸、致残的异质性疾病,包括脊髓前角运动细胞病变、遗传代谢性疾病、神经肌肉接头疾病和各种神经源性骨骼肌损伤等,累及外周神经、神经肌肉接头及骨骼肌,常表现为肌张力低下和肌无力。常见的症状和体征包括呼吸衰竭、骨骼畸形、关节挛缩、步态异常,伴或不伴感觉异常和心脏受累等。新生儿期往往表现为四肢松软、呼吸乏力、关节挛缩和吞咽困难等[1, 2]。大部分NMDs的临床表现非常相似或者不典型,给临床诊断带来困难。详细的病史询问、神经系统体格检查、实验室及诊断性检查有助于疾病的定位。肌肉病理活检(MB)可从蛋白质和病理结构水平认识NMDs的发生及发展,二代基因测序(NGS)技术有助于鉴别特异性的遗传缺陷。目前国内有关新生儿期NMDs的报道多为个案[3-6]。本文回顾性分析复旦大学附属儿科医院(我院)新生儿科收治的连续的确诊NMDs病例的临床资料,旨在加强对新生儿期NMDs的临床认识。
1 方法
1.1 我院新生儿MB及NGS检测临床常规 临床发现有肌张力低下或肌无力的表现(四肢松软、呼吸困难、吸吮无力、多发关节挛缩和吞咽困难等),排除有明确严重感染、甲状腺功能减退症、低血糖、颅内出血和脊髓占位病变等;待病情平稳,完善分子核型筛查,排除染色体结构或数目异常;病情危重,直接行MB及NGS检测。MB医嘱由主治医师决定。
1.2 纳入标准 同时满足以下条件:①2016年1月1日至2018年12月31日我院新生儿科住院病例;②完成MB和NGS检测;③临床发现+MB或NGS确诊NMDs。
1.3 资料截取 从病历资料中截取患儿的临床信息:家族史(近亲结婚,流产、死胎),产前病史(胎动减少、羊水过多及羊水过少),出生史(性别、孕周、出生体重、Apgar评分和分娩方式),新生儿期病史(呼吸支持方式、喂养方式、合并其他畸形等),常规实验室检查结果[磷酸肌酸激酶(CK)、乳酸、血氨],MB病理报告,NGS检测结果及出院情况。
1.4 NGS及阳性结果分类 本文病例样本处理、测序、变异分析和结果解读均参照我院分子诊断中心建立的高通量测序数据分析和临床诊断流程2.0[7]。选用Agilent ClearSeq Inherited Disease试剂盒(包含2 742个已知致病基因)。测序试验采用Illumina Hiseq平台,与人类参考基因组序列比对,98%的目标捕获区域测序深度>20×。变异采用VEP软件(Variant Effect Predictor, Ensembl 73)进行注释。3个主要的收录已知或疑似致病变异的数据库ClinVar、OMIM和HGMD用于筛选已知的致病变异,同时采用多种工具预测错义变异的危害性、注释非编码区序列等。基于人群的大规模测序数据库用于排除在正常人群中具有较高频率的变异。变异使用Clinic Sequence Analyzer(CSA, WuXi NextCODE)进行评估,并依据美国医学遗传学与基因组学学会(ACMG)发布的《序列变异解读标准和指南》对每个变异进行分类。变异按照人类基因组变异协会(HGVS)规则进行命名。阳性变异采用Sanger测序进行验证。
1.5 MB病理检测 无菌操作下,行肱二头肌或股四头肌肌肉活检。将新鲜肌肉组织标本在液氮预冷的异戊烷中固定,冰冻切片。常规进行组织化学染色(HE、改良Gomori三色、ORO、PAS),酶组织化学染色(NADH-TR、SDH、COX、SDH/COX、ATPase、NSE、ACP、AMP等)和免疫组织化学染色(行Alexa Fluor 555和FITC标记分别的dystrophin、α、β、γ、δ-Sarcoglycan、Lamininα2、Caveolin-3、Dysferlin及Collagen Ⅵ等单克隆抗体染色)。
1.6 预后 以患儿出院时是否存活为判断预后的结局指标;因各种因素自动出院的患儿,电话随访患儿的存活状况。
1.7 统计学方法 应用SPSS 20.0软件进行数据处理,非正态分布的计量资料以中位数表示,计数资料以百分比(%)表示。
2 结果
2.1 一般情况 25例NMDs纳入本文分析。男14例,女11例。早产9例,足月16例;顺产10例,剖宫产15例;孕周27+4~41+5(中位数38+3)周,出生体重980~4 050(中位数 3 000)g。住院时间2~105(中位数30)d,行肌肉活检为生后2~114(中位数21)d,行肌肉活检时的纠正胎龄32+1~51+4(中位数41+3)周。
2.2 临床发现 产前检查发现胎动减少2例(8%),羊水过多10例(40%)。特殊面容5例(20%),合并其他畸形6例(24%)。1 min Apgar评分≤7分13/23例(56.5%)。肌张力低下24例(96%)。需呼吸支持22例(88%),其中有创呼吸支持13例(52%),无创呼吸支持9例(36%)。3例禁食状态,需鼻饲喂养20/22例(90.9%),2例经口喂养。神经肌肉相关疾病16例(64%),其中先天性肌病11例、先天性强直性肌营养不良2例、先天性肌营养不良1例、线粒体肌病2例;遗传代谢性疾病4例(16%);神经肌肉接头疾病2例(8%);脊髓前角运动细胞病变2例;心脏疾病1例(表1)。
2.3 实验室检查 2例CK明显升高(例13 和25分别为20 104和4 740 IU·L-1),23例CK正常或轻度升高;5例(例4、11、13、22和24)血氨>200 μmol·L-1,2例(例20和22)血乳酸>10 mmol·L-1。
2.4 MB病理报告和NGS检测结果 MB病理阳性19例(76.0%):先天性肌病10例,强直性肌营养不良、脊髓性肌萎缩症(SMA)、类型待定各2例,先天性肌营养不良、先天性肌无力综合征和线粒体肌病各1例。NGS检测阳性23例(92%):NMDs相关基因14例(56%),其中先天性肌病相关基因9例(MTM13例,RYR1、ACTA1各2例,TTN、NEB各1例),先天性肌营养不良1例(LAMA2),先天性强直性肌营养不良2例(DMPK),线粒体肌病基因2例(mtDNA、CPT2各1例);遗传代谢性疾病相关基因4例(LYST2例、PEX1、MOCS1各1例);SMA 2例(SMN1);先天性肌无力综合征2例(SCN4A、CHRNA1各1例);心脏相关基因1例(KCNH2)。
2.5 预后 死亡15例(60%),存活8例(32%),失访2例(8%)。
死亡患儿中NGS检测阳性13/15例(86.7%):神经肌肉疾病相关基因8例(其中MTM13例,ACTA1、TTN、DMPK、mtDNA和CPT2各1例),LYST2例,SCN4A、SMA和KCNH2各1例。
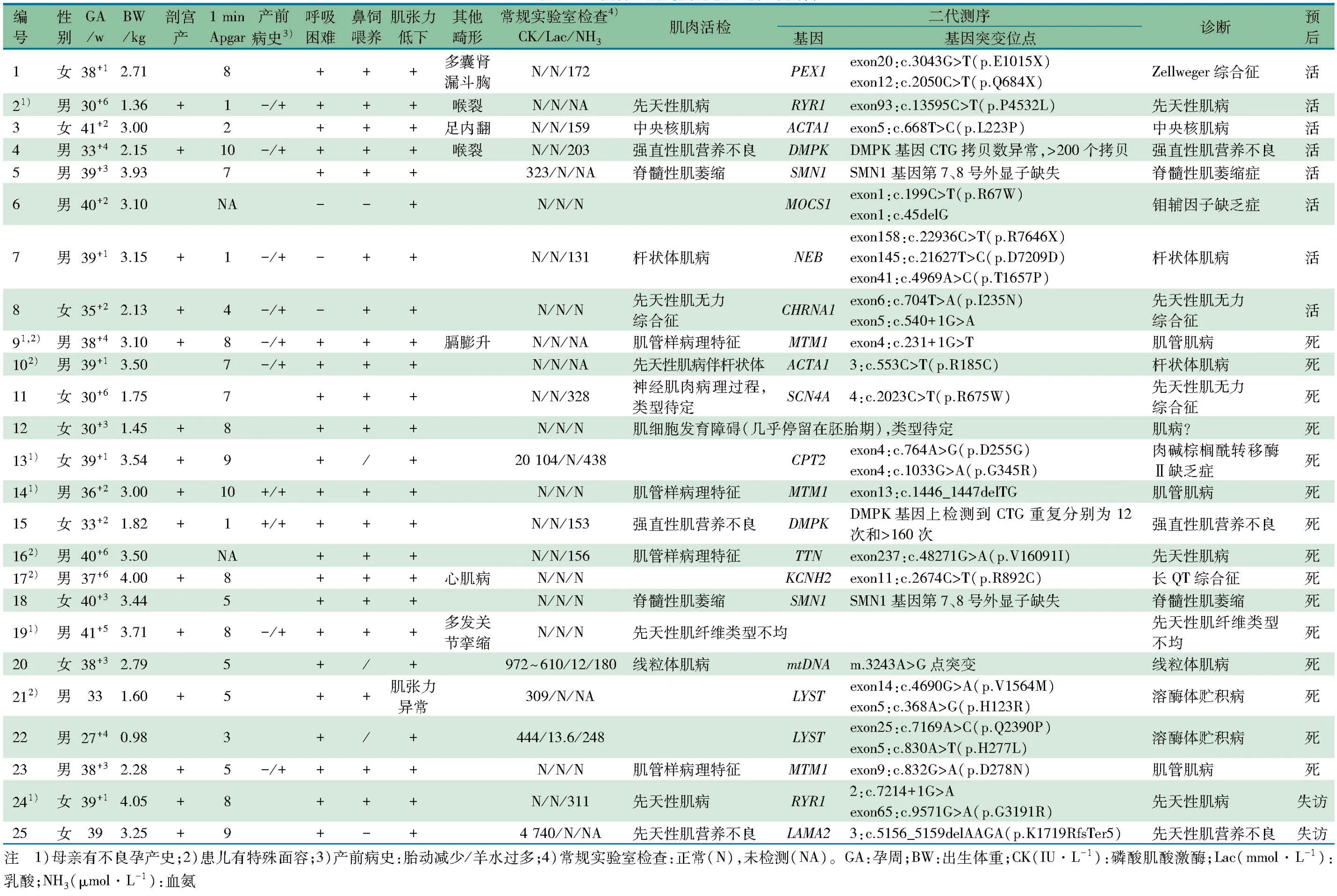
表1 25例经肌肉疾病患儿的临床资料
死亡患儿MB病理阳性11/15例(73.3%):先天性肌病6例,类型待定2例,强直性肌营养不良、SMA和线粒体肌病各1例。
NGS检测和MB病理均阳性9/15例(60%)。2例失访(例24和25),出院时均有氧依赖。例24有不良家族史,系G2P2,第1胎生后20余天因肌张力低下死亡,其MB病理和NGS检测提示先天性肌病(RYR1突变),出院时需鼻饲喂养。例25的MB病理和NGS检测提示先天性肌营养不良(LAMA2突变)。
3 讨论
NMDs是周围神经系统(PNS)异常导致的一组异质性疾病,涉及运动神经元、周围神经、神经肌肉接头或骨骼肌,可能累及中枢神经系统和其他脏器(如心脏、肝脏等)。可于新生儿、儿童或成年期发病,部分疾病呈渐进性过程[1]。脊髓前角运动细胞病变、先天性神经病变、神经肌肉接头疾病、各种类型的肌病及遗传代谢性疾病等均有相似的临床表现,新生儿期起病的临床表现呈非特异性,仅靠临床很难确诊,常易误诊或漏诊,对患儿及其家庭和公共卫生造成极大负担。
本文纳入病例的标准之一为完成MB,Prader-Willi综合征的患儿在新生儿期虽也有相似的临床表现,但通过甲基化-特异性聚合酶链反应(MS-PCR)可确诊,无需常规行MB检查,故予排除。本文包括了如下类型疾病:SMA、神经肌肉接头疾病(先天性肌无力综合征)、神经肌肉相关疾病(先天性肌病、先天性肌营养不良、先天性强直性肌营养不良、线粒体肌病)和遗传代谢性疾病(溶酶体贮积病、过氧化物酶体病、钼辅因子缺乏)。NMDs以神经肌肉相关疾病为主(64%),其次为遗传代谢性疾病(16%)。神经肌肉相关疾病中又以先天性肌病为主(11/16例,68.8%)。本研究例17以呼吸急促起病,临床表现为肌张力低下、吞咽功能差、心功能不全伴心律失常(Ⅰ度房室传导阻滞),心超提示心肌肥厚,由于存在心脏受累,NMDs不能除外,MB未及异常,NGS检测提示KCNH2基因突变(长QT综合征)。
详细的病史询问、体格检查和实验室检查可缩小诊断范围。除外中枢性病因(如胆红素脑病、脑发育畸形等),有如下阳性体征:①对称性、近端或远端肌无力,②面部肌无力或吞咽困难,③原始反射减弱或消失;以及诊断性检查(CK明显升高、肌电图、MB或肌肉MRI成像)有助于定位PNS[2]。新生儿神经系统处于发育阶段,神经系统症状易受多因素干扰,腱反射不易引出,很难区分是否为肌力或肌张力异常,依据体格检查很难进行神经系统的定位和定性诊断,临床工作中更多的是通过临床症状(如惊厥、意识障碍等)来评估是否为中枢或周围性疾病。新生儿期NMDs临床症状缺乏特异性,NMDs在不同时期的临床表现各异。患儿产前检查异常包括胎动减少或消失;由于胎儿吞咽功能受累,常提示羊水过多。新生儿期典型的临床表现为肌张力低下、肌无力(全身性或局灶性)、多发性挛缩(关节挛缩)、呼吸衰竭和吞咽困难,常需呼吸支持及鼻饲喂养,部分病例可有“肌病面容”:面部狭长、长头症、高腭弓及上睑下垂[8]。由于产前呼吸肌运动不良,胸部X线提示肋骨菲薄。先天性肌营养不良实验室检查常提示CK升高[9]。英国[10]的一项单中心研究回顾性分析了125例先天性肌病患儿的自然病程,76%在新生儿期/婴儿期出现临床症状,30.4%出生时需呼吸支持,25.2%需鼻饲喂养。本文发现新生儿期NMDs最常见的临床表现为肌张力低下(96%)、吞咽困难(90.9%)及呼吸困难(88%),排除中枢性、内分泌性及围生期损伤等因素后,当患儿出现上述临床表现时,需高度怀疑NMDs。
MB通过形态学、免疫组织化学和超微结构分析,对病变肌肉做出明确的病理诊断。在先天性肌病中,依据特征性的病理改变进行分型[11]。在无法明确遗传病因时,MB可能会有所帮助。泰国的一项回顾性研究纳入了92例疑似NMDs患儿,MB阳性率为80%,常见的病理类型为肌病(71.6%),无操作相关并发症发生[12]。Maggi等[13]报告了66例先天性肌病的患儿,MB阳性率为81.8%,常见的病理类型是轴空性肌病(54%)。本文MB阳性率76%,常见的病理类型为先天性肌病(52.6%),且无操作相关并发症发生。由于样本例数偏少,无法进行亚型分析。部分NMDs在新生儿期MB无异常表现,当临床高度疑似NMDs,而MB为阴性结果时,常需要NGS来进一步协助诊断。
精准的基因诊断对NMDs患儿的治疗选择、预后评估及家系的遗传咨询均非常重要。MB曾是先天性肌病和先天性肌营养不良诊断的主要手段,随着NGS的日益普及,如果病史和实验室检查结果与基因检测结果一致,可能不再需要MB这种有创性的操作[9]。一些特定的疾病,如线粒体肌病、SMA、强直性肌营养不良和面肩肱型肌营养不良(FSHD),基因检测将取代MB[14]。Maggi等[13]的研究表明66例先天性肌病的患儿,NGS阳性率为66.7%,最常见的基因突变为RYR1(54%)。英国[10]一项回顾性研究分析了125例先天性肌病患儿的自然病程,最常见的致病基因为RYR1(44.4%),突变基因为RYR1、NEB、SEPN1的患儿全部存活;死亡患儿中最常见的突变基因为ACTA1、MTM1、KLHL40。中国[15]409例先天性肌营养不良(CMD)的多中心回顾性调查表明,最常见的基因突变类型为LAMA2(36.4%)。Wang等[16]纳入了214例新生儿肌张力低下患儿,NGS诊断的阳性率为22.5%,NGS结合传统的筛查方法(如染色体核型分析、遗传代谢性疾病的串联质谱分析、Sanger测序、多重连接探针扩增技术及MS-PCR),肌张力低下的确诊率可提高至62.9%。本文NGS检测阳性率为92%,以先天性肌病相关基因为主(39.1%)。
目前诊断NMDs尚无明确的金标准。本研究中MB病理阳性的病例有2例NGS结果阴性(10.5%)。NGS阳性的病例中MB病理阴性为16例(26.1%)。部分代谢性肌病临床早期诊断可尽早采取干预措施,改善预后[1]。MB属于微创操作,可在短期内做出病理诊断,且无操作相关的不良反应。既往基因解读报告所需时间较长。随着3代基因测序技术的开展,可能会缩短基因报告的时间,可先行基因检测[17]。
目前NMDs多无特异性的治疗方法,主要是针对肌无力所引起的并发症给予对症支持治疗。随着医疗技术的发展以及基因治疗的兴起,已有部分神经肌肉疾病如SMA治疗的病例报道。2016年12月美国FDA核准Nusinersen作为治疗儿童和成人SMA的首个药物[18]。目前已有多篇研究表明Nusinersen能改善SMA患儿的临床症状[19, 20]。
本研究不足和局限性:纳入的研究对象为单中心同时行MB及NGS检测的患儿,病例少且来源较单一,肌病分类复杂,无法进行亚组分析。
综上所述,临床高度疑似NMDs的患儿,通过详细的病史询问、体格检查及常规实验室检查,结合MB病理诊断和NGS检测,能极大提高新生儿期NMDs的确诊率。对于因病情危重死亡的患儿来说,明确诊断可能有助于家庭再生育时进行产前咨询。新兴治疗策略的出现将为诊治NMDs带来希望。
