小麦产量相关性状的全基因组关联分析
2020-07-31史雨刚王曙光曹亚萍范绍强孙黛珍
张 东,张 政,史雨刚,王曙光,曹亚萍,范绍强,杨 斌,孙黛珍
(1.山西农业大学农学院,山西太谷 030801;2.山西省农业科学院小麦研究所,山西临汾 041000)
小麦(TriticumaestivumL.)是世界上主要的粮食作物之一, 全世界超过45亿人以其为主食[1]。随着气候的变化、耕地面积的不断减少及人口数量的迅速增长,小麦供需矛盾日趋尖锐,提高小麦品种产量潜力就显得十分重要[2]。穗数、穗粒数、千粒重、株高、穗长等其他农艺性状与小麦产量显著相关[3-5],对这些性状的全基因组进行关联分析,有助于掌握小麦产量形成的遗传信息,促进小麦高产育种。
全基因组关联分析(genome-wide association study,GWAS)由Risch等首先提出[6],是以连锁不平衡为基础,在全基因水平上对自然群体中目标性状的遗传变异与基因多样性进行关联,筛选与表型变异相关分子标记的一种策略,也被称为连锁不平衡作图[7-8]。目前,关于小麦产量相关性状的全基因组关联分析已有一些报道。陈广凤等[9]以205份中国冬小麦品种(系)为材料,利用24 355个SNP标记,在4个环境条件下对农艺性状进行关联分析,发现11个标记至少在2个环境下与株高相关联;邓梅等[10]利用172个产量相关多态性SSR标记, 在繁6及其衍生后代中鉴定出4个与穗长相关联的位点;赵丹阳等[11]利用52份黄淮麦区小麦材料关联到5个与单株穗数显著关联的标记;张彦军等[12]在43份不同来源的和尚头小麦中,利用150对SSR标记关联到2个与穗粒数显著相关位点;张国华等[13]以128份黄淮麦区小麦品种(系)为材料, 在4个环境下检测到13个与千粒重显著关联的位点。但是由于小麦的产量相关性状是由多基因控制的数量性状,且受环境的影响较大,所以有必要利用多个群体在多个环境下进行关联分析,以便找到稳定的关联位点用于育种。本研究以 236 份小麦种质资源构成的自然群体为材料,利用均匀分布于小麦 21 条染色体上的 106对SSR 标记,对小麦单株穗数、主穗粒数、千粒重、株高、穗长5个产量相关性状在2年4个环境下进行产量相关性状的全基因组关联分析,筛选优异等位变异,为分子标记辅助选择育种提供依据。
1 材料与方法
1.1 试验材料与处理
236份种质资源包括中国冬麦区推广的小麦育成品种、骨干亲本。其中,有225份来自于中国,1份来自智利,4份来自意大利,6份未知来源材料。
236份材料于2016年9月至2017年7月及2017年9月至2018年7月种植在山西农业大学农作站(北纬37°25″,东经112°25″)。试验用随机区组设计。每一份材料种植一行,每行点播70粒种子,行长2.0 m,行距0.2 m,株距3.0 cm,行向南北。设置雨养和灌溉两种水分管理方式,各进行3个重复,每一重复生长条件除水分管理方式之外均相同。雨养条件下不进行人工灌溉,小麦生长仅依靠自然降雨,而灌溉条件下分别在越冬前期、返青期、拔节期和灌浆中期进行人工灌溉,以满足小麦在生长发育过程中对水分的需要。
小麦成熟之后,每个重复随机从中选取5株生长健壮、整齐一致的植株,考察株高、穗长、单株穗数、主穗粒数和千粒重,以其平均值作为性状的观测值。
1.2 基因组DNA提取
每个材料于培养皿中培养至二叶一心期(25 ℃,12 h光照;20 ℃,12 h黑暗),取约0.2 g小麦叶片,研磨之后,使用CTAB法提取小麦叶片中的DNA,ddH2O溶解之后进行琼脂糖凝胶电泳检测DNA质量。
1.3 SSR标记的筛选
根据标记的多态性及PCR扩增的稳定性,从大量的SSR标记中筛选出106对SSR标记,其分布见表1。
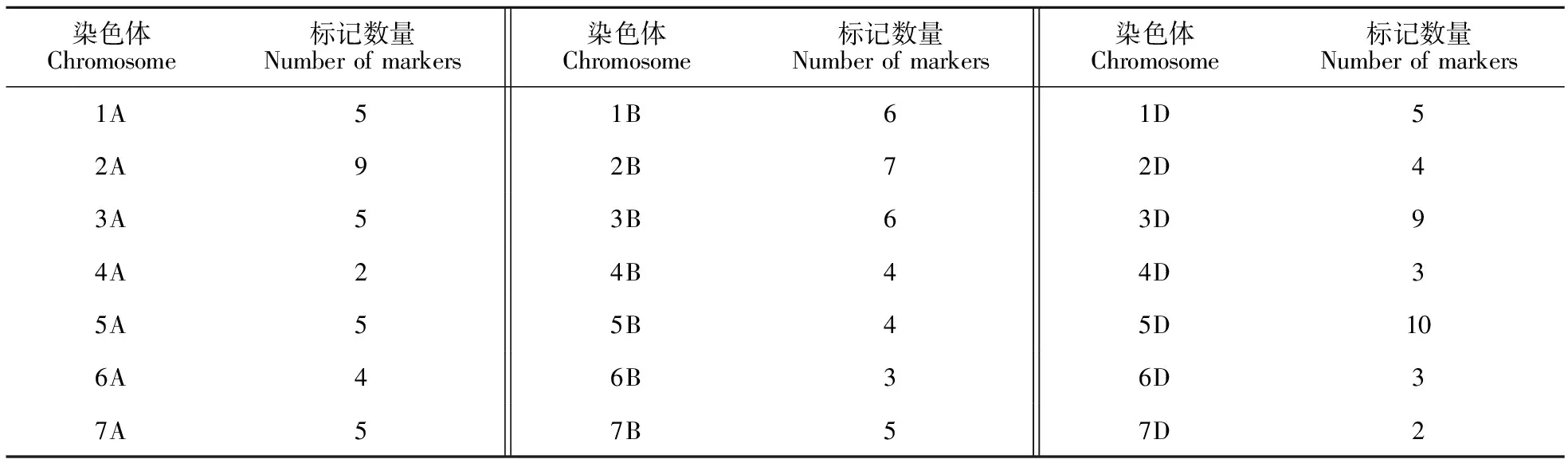
表1 106对SSR标记在染色体上的分布
PCR程序为95 ℃预变性5 min;95 ℃变性30 s,退火30 s,72 ℃延伸45 s,运行34个循环;72 ℃延伸10 min;10 ℃保存。使用Taq酶(购置于天根生化科技有限公司)进行PCR扩增,引物由生工生物工程(上海)股份有限公司合成,反应总体系为15 μL,0.3 μL前引物,0.3 μL后引物,1 μL模板DNA,1.5 μL buffer,1.2 μL dNTPs,0.15 μL Taq酶,10.55 μL ddH2O。为防止PCR产物中残留的盐和蛋白影响毛细管电泳检测,对扩增产物进行纯化,利用ABI3730 DNA测序仪(Applied Biosystems,Foster City,USA)检测扩增产物的片段大小;使用GenemapperV3.7软件进行数据读取。
1.4 统计分析
依据Liu等[14]的方法对每个SSR位点的等位变异数、等位基因频率、多态性信息指数(PIC)应用PowerMarker V3.25软件进行计算。使用张政等[15]的方法应用Structure V2.3.2软件,利用上述的106对SSR标记进行群体结构分析,每个K值重复5次,取值范围设定为1~15,将MCMC(Markov Chain Monte Carlo)开始时的不作数迭代(length of burn-in period)设为50 000,再将不作数迭代后的MCMC设为100 000。亚群的数量依据Evanno等[16]方法用K+Q的模型进行确定;品种间的亲缘系数依据Hardy等[17]的方法使用SPAGeDi软件计算。表型数据依据Bernardo等[18]的方法使用SPSS 20.0软件进行处理。关联分析使用TASSEL 3软件中的混合线性模型(MLM),用群体结构(Q)和亲缘关系(K)为协变量,对标记和表型数据进行关联分析,当P<0.01时为显著关联,当P<0.001时为极显著关联。通过各位点每个等位变异的效应与该位点平均效应值的比较计算等位变异的遗传效应,再通过对等位变异表型效应的解析筛选优异等位变异[19]。
2 结果与分析
2.1 不同环境下小麦农艺性状的表现
两年四个环境下小麦株高、穗长、单株穗数、主穗粒数、千粒重变异广泛,除了单株穗数之外其他性状的变异系数均大于10%(表2)。说明这些性状在品种间的遗传变异性比较大。
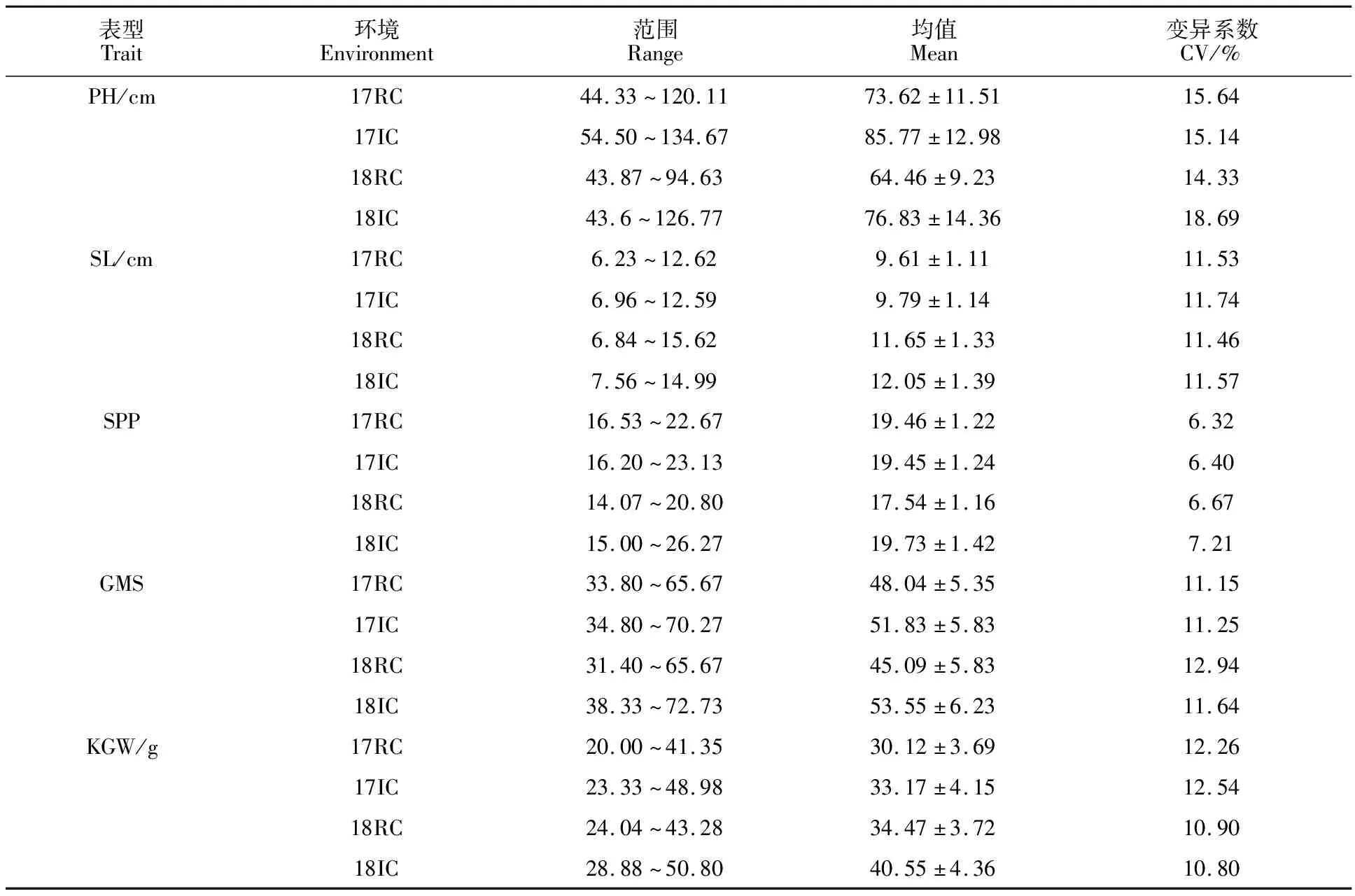
表2 不同环境下产量相关性状的表型变异统计
2.2 利用分子标记的检测结果
用106对SSR标记在236份小麦材料中共检测到874个等位变异,每对标记平均为8.24个,变化范围为2~23个;主要等位变异频率(MAF)的变化范围0.177~0.987,平均为 0.545,其中频率最小的分子标记是gwm268,最大的是cfd106;多态性信息含量(PIC)的变化范围为0.026~0.895,平均为0.550,频率最小的分子标记是cfd106,最大的是gwm174,多态性达到高度水平(PIC>0.5)(表3)。这表明这些小麦品种在分子上的遗传多样性水平较高。
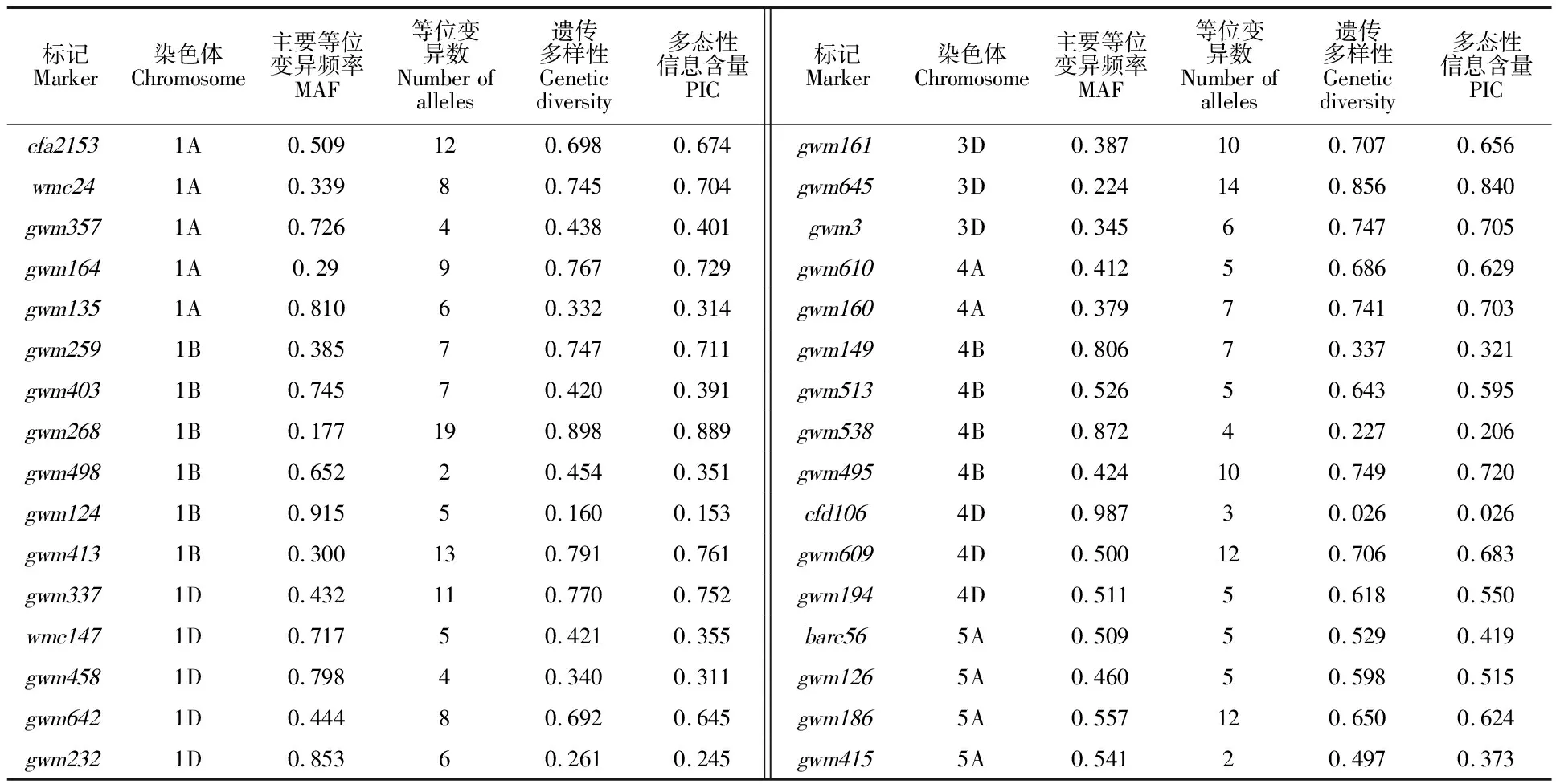
表3 106对标记的遗传多样性数据
2.3 群体结构分析
基于混合线性模型对供试的236个小麦种质资源进行群体结构分析,以设定的K所对应的最大似然值为目标来选择合适的群体数目,计算每个K值下的△K值,根据计算出的△K值绘制ΔK值随K值变化的折线图。K=2时,△K值出现明显的最大值,随后下降(图1),因而选定K=2为最佳的群体数目,将该群体分为两个亚群(图2)。第一亚群包含127份材料(53.81%),第二亚群包含109份材料(46.19%)。其中来源于江苏的63份材料主要分布于第二亚群(76.19%),河南的29份材料主要分布于第一亚群(82.86%),陕西的26份材料主要位于第一亚群(73.08%),山东的21份材料中有19份分布于第一亚群(90.48%),说明来源于上述地区的材料群体结构较为单一。
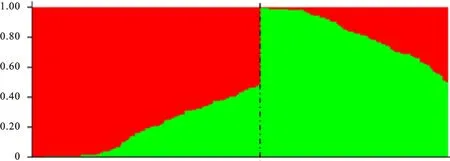
图2 通过Structure V2.3.2软件分析的遗传结构(红色:第一亚群;黄色:第二亚群)
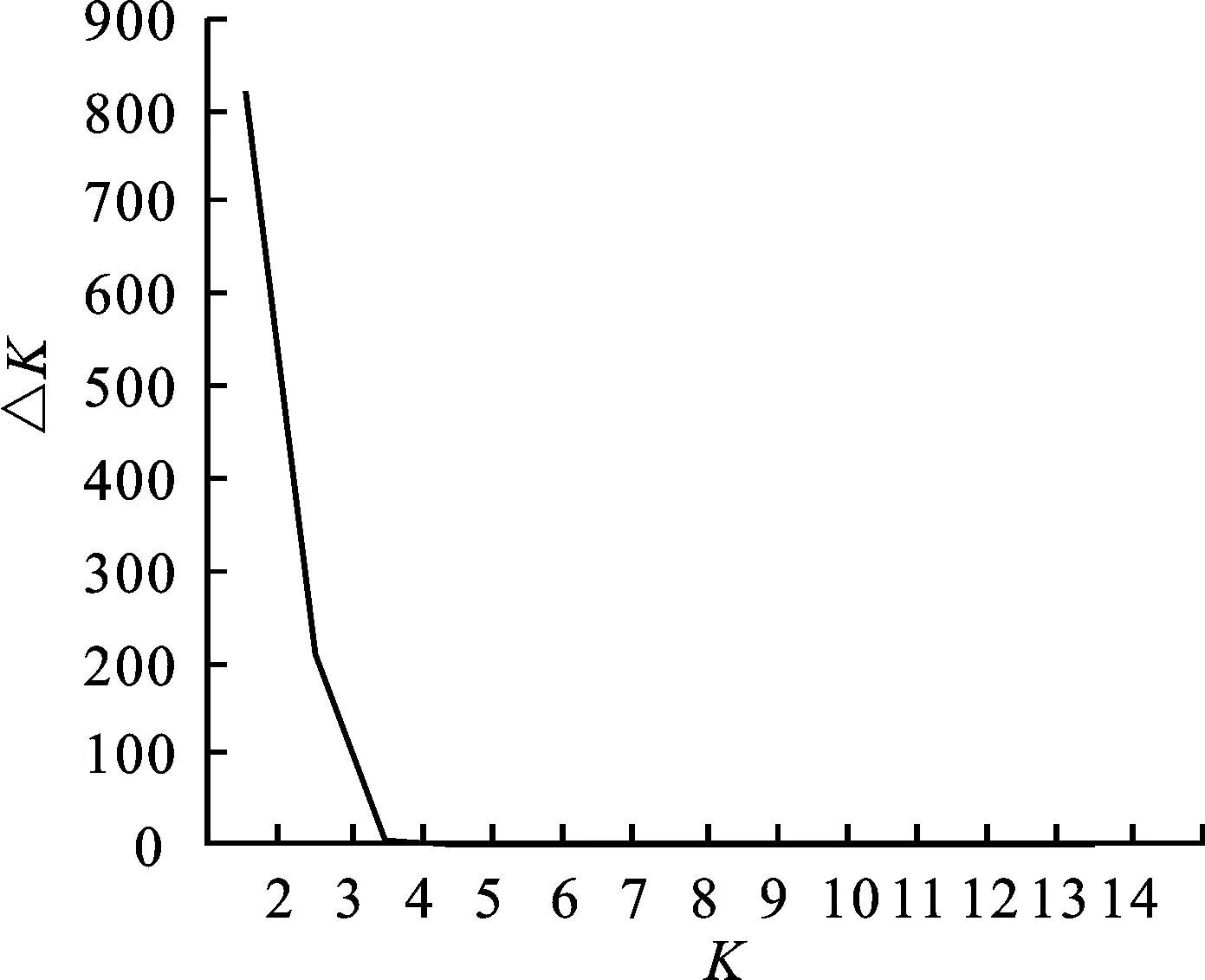
图1 通过假定不同K值计算得到的△K值
2.4 亲缘关系分析
对236份小麦材料之间亲缘关系分析的结果表明,75.62%的品种的亲缘系数小于0.05,其中包括约55.35%材料的亲缘关系值为0,12.57%的材料介于0.05~0.10范围,6.21%的材料介于0.10~0.15范围,2.89%的材料介于0.15~0.20范围,1.22%的材料介于 0.25~0.30范围,0.28%的材料介于0.30~0.35范围,0.14%的材料介于0.35~0.04范围,0.13%的材料介于 0.40~0.50范围,0.09%的材料介于0.45~0.05范围(图3),说明大部分品种(系)之间没有亲缘关系或亲缘关系较远,对关联分析影响较小。
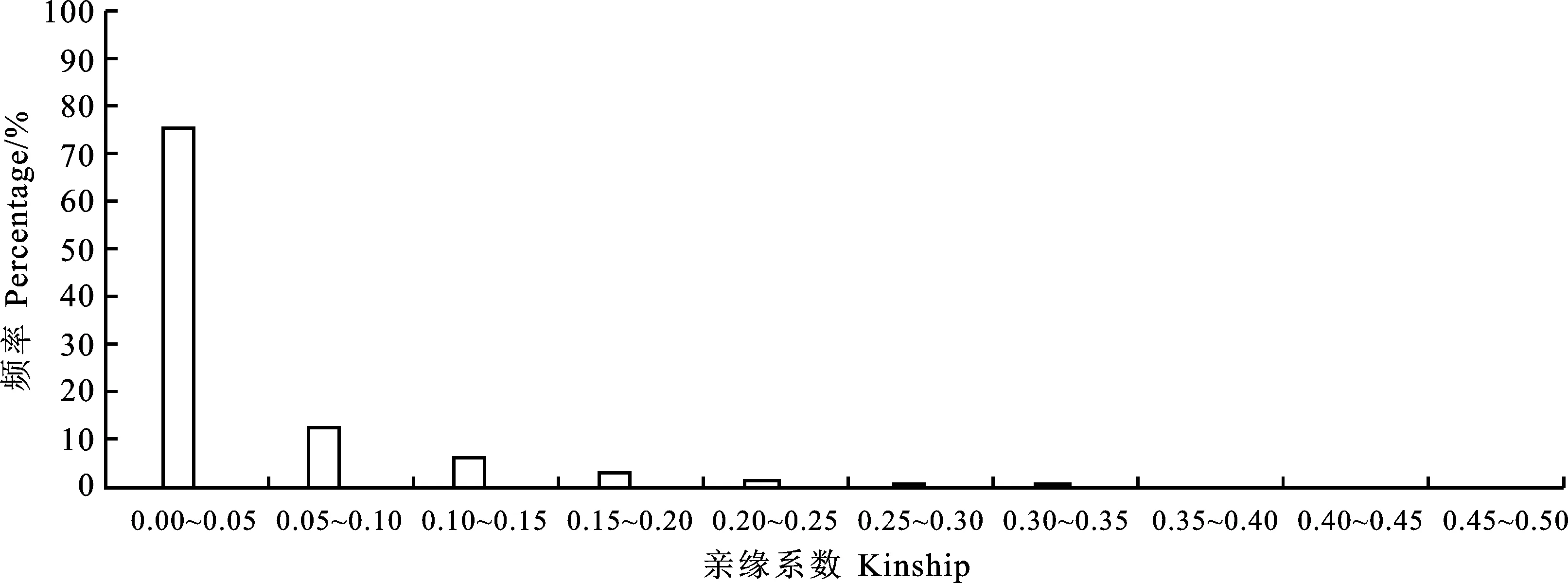
图3 236份小麦品种间亲缘关系的分布
2.5 产量相关性状与标记间的关联分析
经关联分析,在供试材料检测到的位点中,7个位点与株高显著关联(P<0.01),它们分布在1A、5D和6D染色体上(表4)。其中,位于1A染色体上的Xgwm164(1A)在4个环境下均与株高显著关联,表型解释率(R2)为10.55%~ 13.74%;Xgwm55(6D)在2个环境下与株高显著关联,表型解释率分别为11.62%和12.17%。有4个位点与穗长显著关联,它们分布在2B、6B和6D染色体上,表型解释率为11.50%~ 15.65%。其中,Xgwm429(2B)在2个环境下与穗长显著关联,表型解释率分别为11.50%和 15.65%。有7个位点与单株穗数显著关联,它们分布在1A、1D、4D、5B、5D和6B染色体上,表型解释率为7.36%~10.24%。其中,Xwmc415(5B)在2个环境下与单株穗数显著关联,表型解释率分别为8.72%和9.37%;Xgwm642(1D)的表型解释率大于10%。有5个位点与主穗粒数显著关联,它们分布在1D、2D、5D和6B染色体上,表型解释率为6.25%~13.99%,其中Xgwm102(2D)和Xgwm219(6B)的表型解释率大于10%。有3个位点与千粒重显著关联,它们分布在2B、4A和5A染色体上,表型解释率为 7.70%~18.97%,其中Xgwm120(2B)和Xgwm186(5A)的表型解释率大于10%,Xgwm120(2B)的表型解释率最大,达到18.97%。
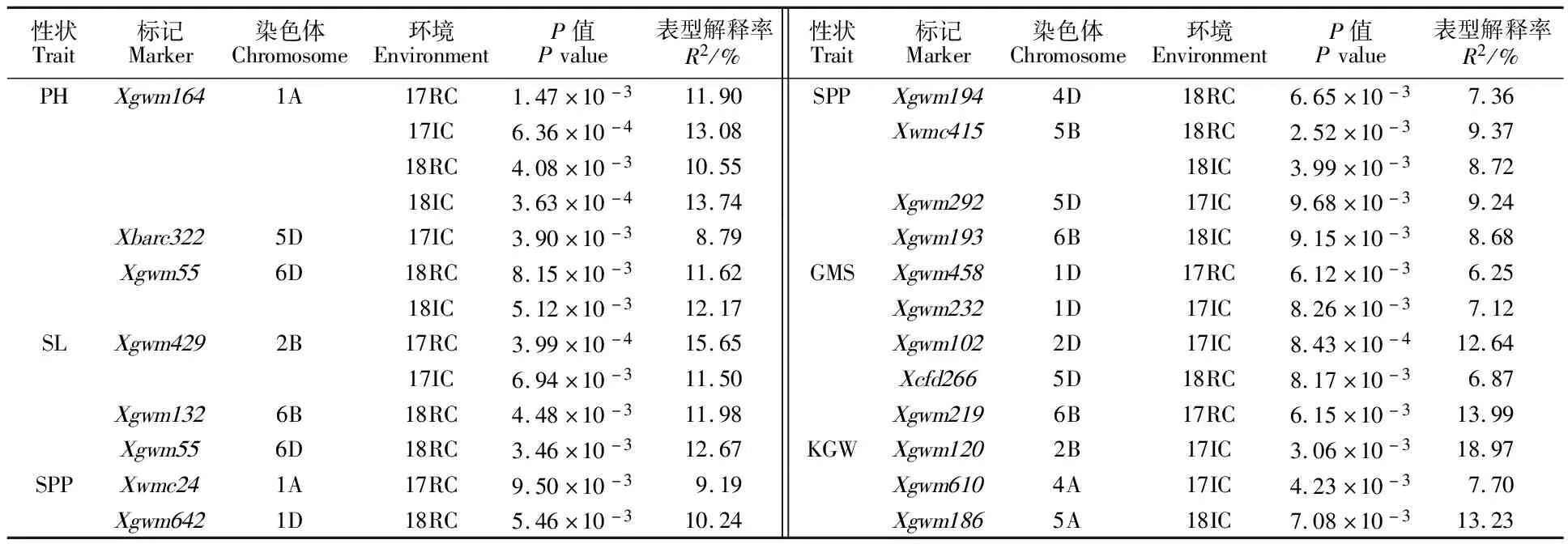
表4 小麦产量相关性状的关联分析
2.6 优异等位变异及其效应
与株高关联的优异等位变异中,Xgwm164-1A118效应最大,可降低株高4.24 cm。与穗长关联的优异等位变异中,Xgwm429-2B207效应最大,可增加穗长0.75 cm。与单株穗数关联的优异等位变异中,Xgwm194-4D129、Xwmc415-5B154、Xgwm292-5D208和Xgwm193-6B166的等位变异频率分别为50.24%、71.71%、70.73%和74.63%,这些位点在育种过程中可能经过较强人工选择,其中Xwmc415-5B154效应最大,可增加单株穗数1.07个。与主穗粒数关联的优异等位变异中,Xgwm232-1D138效应最大,可增加主穗粒数1.93粒;优异等位变异Xcfd266-5D165的等位变异频率为 50.24%。与千粒重关联的优异等位变异中,Xgwm610-4A170效应最大,可增加千粒重 0.92 g(表5)。
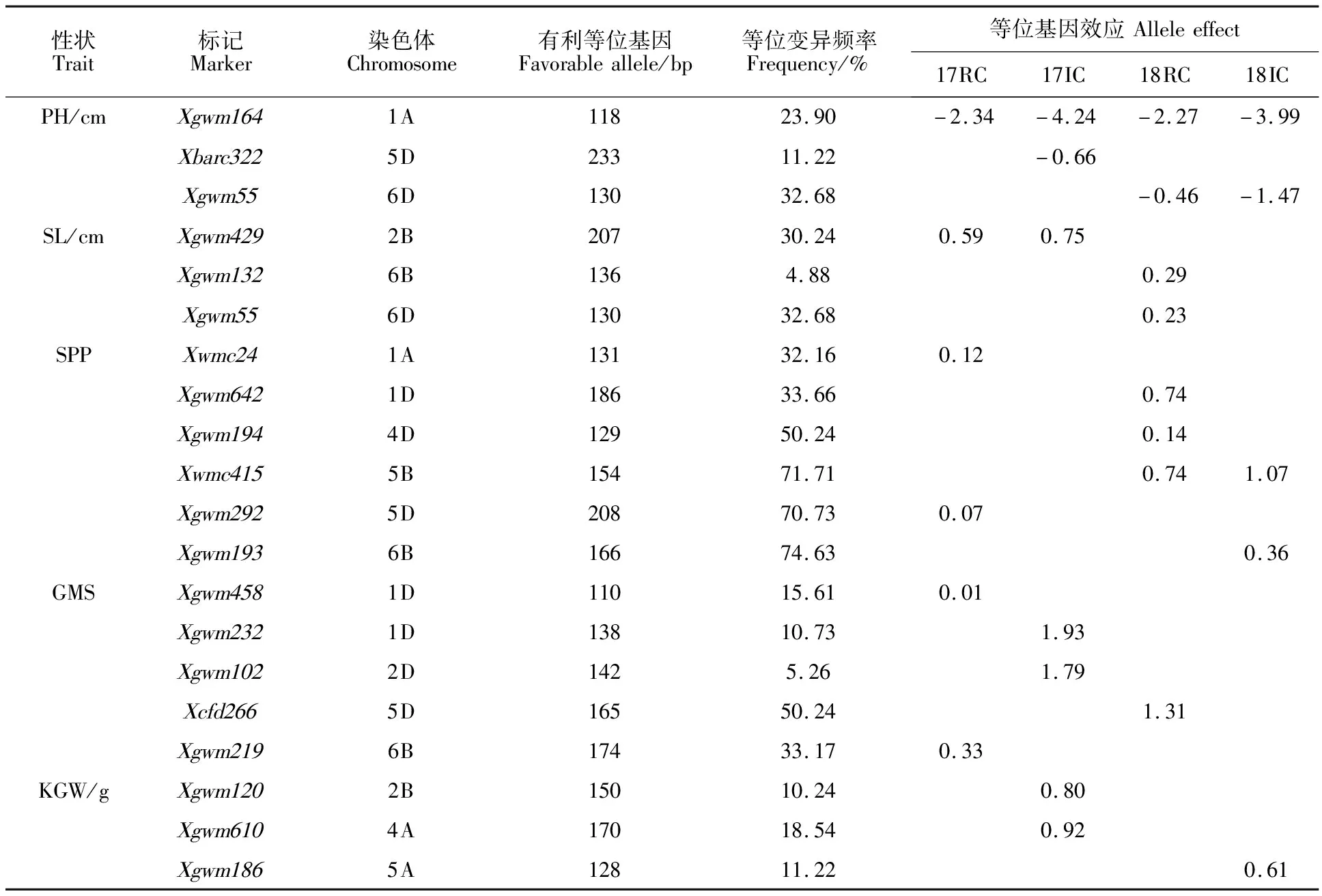
表5 关联标记的优异等位变异及其效应
3 讨 论
影响关联分析结果的主要因素包括表型测量的准确性、群体结构和亲缘关系、标记的遗传多样性、关联分析所选择模型的检测效力等。本研究在两年四个环境下进行,且每个处理均设有三次重复,表型测量时通过随机选5株的方法来减少试验误差。群体结构和亲缘关系是导致关联结果出现假阳性的两个主要因素,假阳性可能带来虚假的等位基因多样性信息,最终导致基因型数据精确度受损。本试验使用Structure V2.3.2软件进行群体结构分析,将供试群体分为2个亚群,使用SPAGeDi软件计算了群体间的亲缘关系,同时将群体结构分析得到的Q矩阵和亲缘关系分析得到的K矩阵作为协变量参与关联分析,以消除由于供试材料的群体结构和亲缘关系所引起的伪关联。所选标记的遗传多样性较高时,可以提高其作图能力,并且增加可供检测的等位基因数量。本研究利用106对SSR标记扫描236份小麦材料,发现这些材料多态性信息指数(PIC)的变化范围为0.026~0.895,平均为 0.550,表明这些小麦品种在分子水平上有较高的遗传多 样性。
目前,已命名的小麦矮秆(Rht)基因有30个,分布于小麦2AS、2BL、2DS、3BS、4BS、4DS、5AL、5DL、6A、7A和7B染色体上[20-23]。本研究发现,位于1A染色体上的标记Xgwm164和6D染色体上的标记Xgwm55分别在4个和2个环境下与株高显著关联。虽然余马等[24]在1A染色体的标记区间Xgwm163-Xpsr11检测到一个与株高相关的QTL,但与标记Xgwm164相距较远,所以推断本研究检测到的这两个与株高相关联的标记位点是新位点。此外,陈晓杰[25]以90份中国冬小麦为材料发现Xgwm164(1A)与小穗数、胞间CO2浓度显著关联,并可增加0.099 3个小穗数、增加0.0919 μL·L-1胞间CO2浓度。Iqbal Saeed[26]对59份冬小麦材料研究发现,Xgwm164(1A)与净光合速率在2个环境下显著关联,表型解释率12.68%。而本研究发现,优异等位变异Xgwm164-1A118可以降低株高4.24 cm,因此该位点具有一因多效性。
其次,本研究发现6D染色体上的标记Xgwm55同时与株高和穗长两个性状显著关联,且优异等位变异Xgwm55-6D130可降低株高1.47 cm,增加穗长0.23 cm;而郭杰[19]研究发现Xgwm55与穗基部第三个小穗结实粒数显著关联,且可增加基部小穗结实数0.12粒;Perretant等[27]利用旱选10号和鲁麦14构建的双单倍体(DH)群体中定位到一个微效的籽粒硬度QTL位点与Xgwm55-6D连锁,所以该位点也具有一因多效性。另外,本研究在2个环境下均检测到标记Xwmc415(5B)与单株穗数显著关联,且优异等位变异Xwmc415-5B154,可以增加单株穗数1.07个。而杨林等[28]利用中国春/兰考大粒的F2群体在5B染色体的标记区间上Wms499-Wms371定位到一个与单株穗数相关的QTL,与标记Xwmc415(5B)相距仅4 cM;张冬玲[29]研究发现Xwmc415与千粒重、穗粒数显著关联,且优异等位变异Xwmc415-5B154对千粒重和穗粒数均有显著的增效作用。显然,以上三个标记位点对于产量因子的作用都是正向的,因此利用这三个标记位点进行辅助选择矮秆小麦品种的同时,还可增加每穗小穗数、改善胞间CO2浓度或者增加穗长和基部小穗结实数,改善品质或者可同时改善单株穗数、千粒重和穗粒数。
另一方面,已经在染色体1A、1B、2A、2B、2D、3A、3B、4A、4B、5A、5B、5D、6A、6B、7A和7B染色体上检测到与穗长相关的QTL[30-33]。本研究中,在2017年雨养与灌溉两种环境下,检测到2B染色体上的标记Xgwm429与穗长相关联,优异等位变异Xgwm429-2B207可以增加穗长0.75 cm,与侯立江[30]在2B染色体标记区间Xwmc764-Xwmc770中检测到的QTL相距仅3 cM,可能为同一位点。同时郭杰[19]研究发现Xgwm429可同时提高顶部第一个小穗位的结实粒数0.02粒,然而付必胜等[34]检测到标记Xgwm429在3个环境中与赤霉病抗性相关,可增加0.3个感赤霉病小穗数。因此,在育种过程中利用该标记进行辅助选择时,一定要考虑到对赤霉病的感病性。
