超高效液相色谱-串联质谱法测定葡萄中16种植物生长调节剂的残留量
2020-07-29占绣萍陈建波黄兰淇
占绣萍,陈建波,马 琳,陈 秀,黄兰淇,刘 彬
(1.上海市农业技术推广服务中心,农业农村部农药质量监督检验测试中心(上海),上海 201103;2. 上海市青浦区蔬菜技术推广站,上海 201200)
植物生长调节剂(plant growth regulator,PGRS)是人工合成的对植物的生长发育有调节作用的化学物质,具有和植物内源激素具有相似生理功效和相似化学结构的化学合成物。植物生长调节剂能够有效的掌控植物的生长和加强其抗逆性,有效的解决了许多农业生产中常见的技术问题,展现出了高效的调控能力和广阔的应用前景。植物生长调节剂也是属于农药的一种,虽然大部分毒性较低,但是长期食用含有PGRS的食品,会对人体健康造成潜在的健康危害[1-5]。我国在新版GB 2763-2019《食品中安全国家标准 食品中农药最大残留限量中》[6],已经制定了19种植物生长调节剂在某些农产品中的最大残留限量标准,可见安全使用植物生长调节剂越来越多的得到了政府的重视和人们的关注。
目前,国内外关于植物生长调剂的检测方法主要采用酶联免疫法、毛细管电泳法、气相色谱法、气相色谱-质谱法、液相色谱法、离子色谱法、液相色谱-质谱联用法[7-19]。其中液相色谱-质谱联用以其高灵敏度和高选择性,被更多的应用。样品前处理方法中,分散固相萃取技术自2003年提出以来,在水果、蔬菜等农产品农药多残留检测中得到广泛应用。其优点是前处理简单方便,不仅化学试剂用量少,还能有效去除色素、果糖、脂肪等物质的干扰,不易造成待测成分损失。本研究采用缓冲盐/分散固相萃取和液相色谱-串联质谱技术,兼具简单、快速、高灵敏度、抗干扰能力强等优点,适合葡萄中植物生长调节剂多残留的定性定量检测。
1 实验部分
1.1 主要仪器与装置 Agilent1290 UPLC-Agilent6460超高效液相色谱-三重四极杆串联质谱联用仪:配ESI源,T25DS25高速匀浆分散器;GB11240-89电热恒温水浴锅;AC-40氮吹浓缩器;国华SHAC恒温振荡器;TDL-5-A离心机;Milli Q超纯水系统;漩涡混合器(SCI LoGex MX-S)。
1.2 试剂乙腈和甲醇 (色谱纯,德国Merck),甲酸(色谱纯,美国Fluka),乙酸铵(色谱纯,美国TEDIA),高纯水,氯化钠(分析纯);十八烷基键和硅胶(C18)吸附剂:43~60μm,N-丙基乙二胺(PSA)吸附剂:40~60μm,无水MgSO4, 净化管(含100 mg PSA+300 mg MgSO4)。16种农药标准品: 矮壮素、甲哌啶、抑芽丹、N6-腺嘌呤、赤霉酸、苄基腺嘌呤、吲哚乙酸、脱落酸、噻苯隆、对氯吡氧乙酸、氯吡脲、芸苔素内酯、多效唑、2,4-D、烯效唑、戊唑醇(纯度均>96%,德国Dr.Ehrenstorfer GmbH)。
称取适量标准品,分别用甲醇配制成100mg/L的储备液并储存在-4℃冰箱中。根据实验需要用甲醇稀释标准储备液,配成适当浓度的混合标准工作溶液。
1.3 样品前处理方法
1.3.1 提取 准确称取粉碎并混匀后的葡萄样品10g (精确至0.01g),置于50mL聚四氟乙烯离心管中,加入20mL5%乙酸的乙腈溶液,高速匀浆提取1min,加入4g硫酸镁、1g柠檬酸钠、0.5g柠檬酸氢二钠、1颗玻璃均质子,涡旋1min,4 000r/min离心5min,静置60min,待乙腈相和水相分层。
1.3.2 净化 葡萄样品净化:取4mL上层乙腈相溶液置于QuEChERS离心管(含100mg PSA+300mg MgSO4),涡旋混合1min,以4 000r/min 离心5min,移取上清液,过0.22μm有机滤膜后,待测。
1.4 检测条件
1.4.1 色谱条件 色谱柱: Agielnt Poroshell 120 EC-C18柱( 3.0mm×100mm,2.7μm) ; 以0.1%甲酸+5mmoL乙酸铵水溶液( A 相) 和甲醇(B 相) 为流动相进行线性梯度洗脱; 梯度洗脱程序: 0~1.0min,A 相的比例保持90%,1.0~2.5min,A 相的比例由90%降到70%,2.5~4.5min,A 相的比例由50%降到5%,4.5~7.0min,A 相的比例由5%降到1%,7.0~8.0min,A 相的比例保持1%%; 流速为0.35mL/min; 进样量: 2 μL; 柱温: 40 ℃。
1.4.2 质谱条件 采用AJS ESI源,分段多反应监测模式(MRM)检测(其中第1段:0~2.30min,正离子扫描,不加EMV电压;第2段:2.3~4.8min,正离子扫描EMV加400v;第3段:4.8~5.4min,负离子离子扫描EMV加400v; 第4段:5.4~8.0min,正负离子同时扫描,负离子EMV加400v,正离子不加EMV)。雾化气压力:310.3kPa(40psi);干燥气温度与流速:300℃,7L/min;鞘气温度与流速:350℃,12L/min;毛细管电压:正离子3 000v,负离子3 500V。16种农药的定量和定性离子、碰撞能量和碎裂电压等参数(表1)。
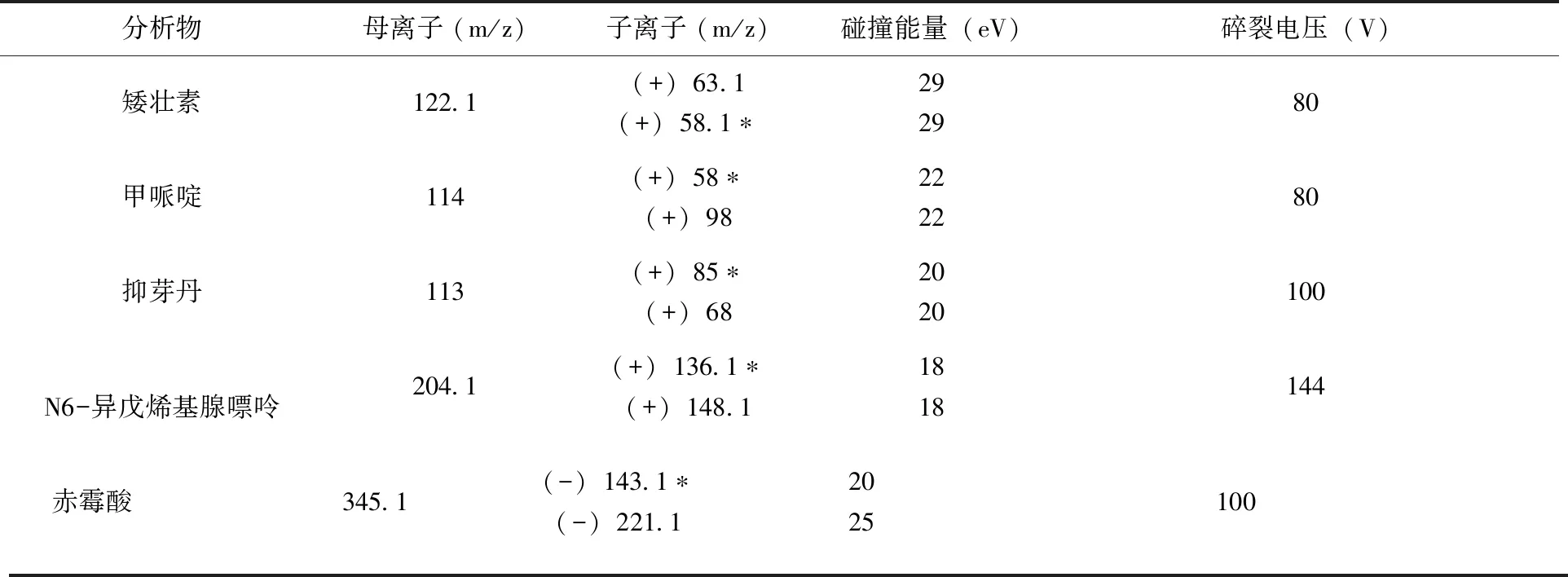
表1 16种农药的质谱分析参数
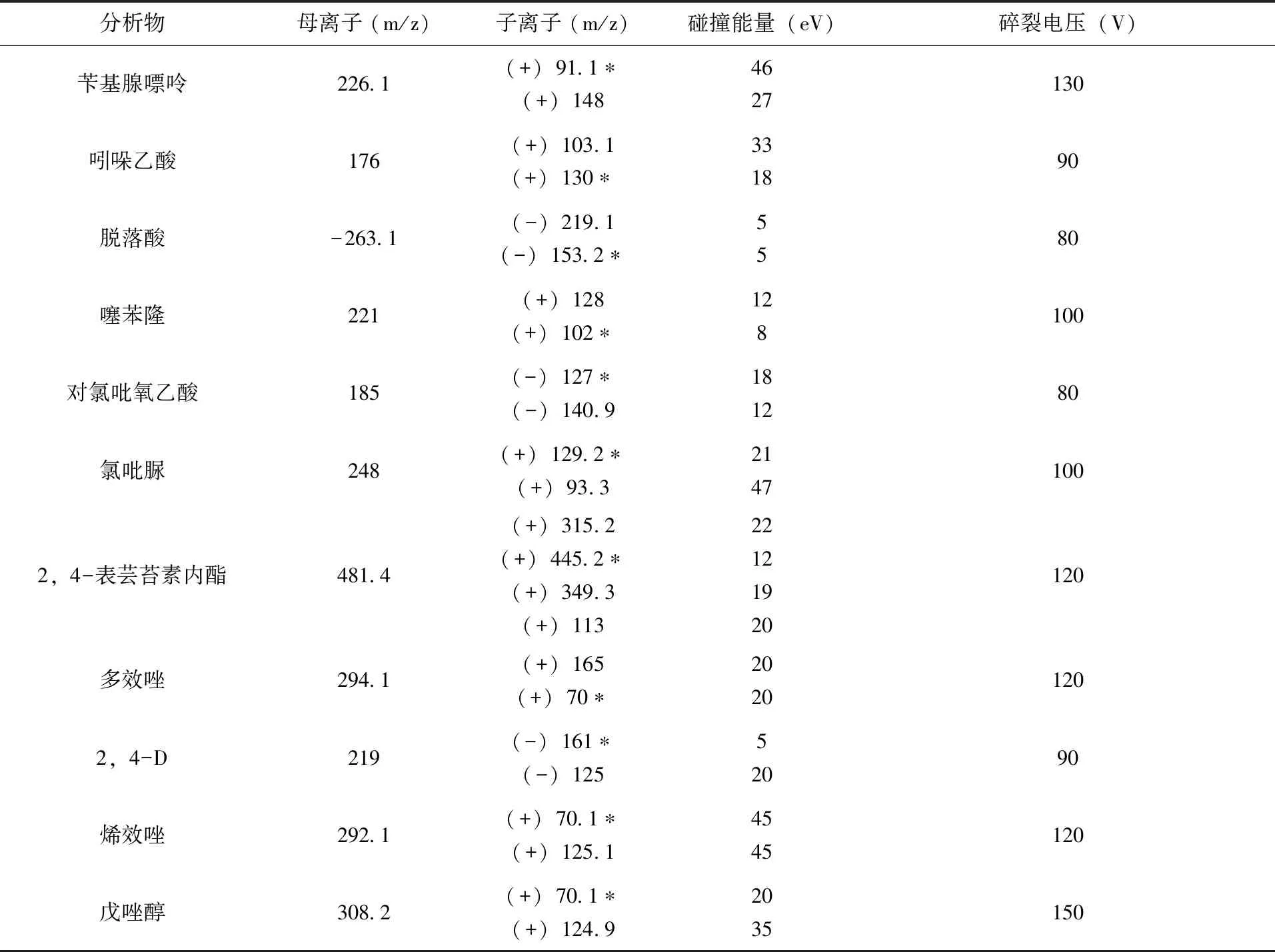
续表
2 结果与讨论
2.1 色谱条件优化 本试验采取正负切换离子模式采集,选择流动相时,不仅需要关注正离子电离化合物的相应,更重要的是负离子电离化合物在流动相影响下的峰形和相应灵敏度情况。试验选择了有机溶剂甲醇、乙腈作为强洗脱相;水、含0.1%甲酸溶液、含5mmol/L 甲酸铵、含5mmol/L乙酸铵溶液作为弱洗脱相,对其不同搭配进行了比较。
首先以甲醇-水、乙腈-水作为流动相来分析,结果发现在乙腈-水洗脱的时候,副模式下电离的化合物如赤霉酸、脱落酸、对氯吡氧乙酸及2,4-D灵敏度很低,几乎不出峰;且正模式电离的化合物如苄基腺嘌呤、吲哚乙酸、氯吡脲、噻苯隆、N6-异戊烯基腺嘌呤等5种灵敏度偏低的化合物,响应也明显偏低;其他灵敏度好的化合物,虽然用乙腈电离被增强,但是已经超出了检测必需的必要度。所以本试验选择甲醇作为强洗脱相。对比甲醇-水、甲醇-0.1%甲酸水、甲醇-0.1%甲酸-5 mmoL/L乙酸铵溶液、甲醇-0.1%甲酸-5 mmoL/L甲酸铵溶液和乙腈-0.1%甲酸-5 mmoL/L乙酸铵溶液为流动相时,发现甲醇- 0.1%甲酸-5 mmoL/L乙酸铵溶液对于灵敏度偏低的化合物电离效果最好,响应高,对副模式采集的峰形和响应都较好。因此选择甲醇和0.1%甲酸-5 mmoL/L乙酸铵溶液作为流动相。
2.2 质谱条件优化 使用单针自动进样分析目标化合物的标准溶液,优化16种农药的质谱条件。化合物进入一级质谱后,可产生稳定的[M+H+离子或[M-H]-离子。其中对氯吡氧乙酸、2,4-D、脱落酸、赤霉酸,形成稳定的[M-H]-离子,其他农药均选择了[M+H]+离子。确定了化合物母离子后,在SIM 模式下,对化合物的Fragment(碎裂电压)进行优化;母离子进入二级质谱,发生断裂或重排等反应产生不同的离子碎片。在Product Ion模式下,对化合物母离子施加一定量的碰撞能量(CE),得到其相应的离子碎片;最后在MRM 模式下,优化目标物离子碎片的最佳碰撞能量。对于正负离子同时采集时,发现负离子灵敏度偏低,根据化合物灵敏度高低不同,故EMV(电子倍增管电压)负离子扫描时分别加-200、-400 V,而正离子采集的化合物中,仅抑芽丹灵敏度较低,所以分段采集并给它加+200V,从而提高检测灵敏度。并同时优化了质谱毛细管电压、碎裂电压、碰撞能量、电子倍增管电压等质谱条件。16种农药和空白葡萄的MRM 色谱图(图1、图2)。
2.3 前处理条件的优化
2.3.1 提取溶剂优化 查询文献,大多数植调剂在酸性条件下提取,稳定性明显高于中性条件的提取。因此本研究采取乙腈加不同浓度的乙酸作为提取剂,来比较回收率情况。以0.1%、1%、2%、5%乙酸乙腈溶液作为提取溶剂,平行操作3次,考察4种提取溶剂在同一种净化包(100mg C18、100mg PSA 、300mg MgSO4混合)处理效果(图3) 。当提取溶剂为5%乙酸乙腈时,除了矮壮素、甲哌啶外,其他植调剂平均回收率70.4%~109.7%,而且明显优于其他三种提取剂。这可能是由于酸性条件抑制了植物生长调节剂中羧基的电离,从而增加了其在乙腈中的溶解度。
2.3.2 净化条件优化 QuEChERS方法作为一种新型的前处理方法,较普通的SPE方法具有明显的优势;用QuEChERS方法溶剂使用量相对较少,无需淋洗,操作简单,所需时间少,所需净化剂的材料价格相对低廉。如N-丙基乙二胺(PSA)、C18、石墨化炭黑(Carb)是常用的吸附剂材料。PSA吸附剂可有效去除样品中的有机酸、脂肪酸和糖等干扰物;C18吸附剂对极性较弱的脂肪酸、烯烃类及甾醇类、色素等大分子基质干扰物有较好的吸附效果;Carb吸附剂对去除叶绿素类胡萝卜素等色素以及固醇类杂质有很好的效果。
试验选择葡萄样品进行研究,C18和PSA均能不同程度的去除花色苷,但是比较提取溶剂优化试验中发现:去除杂质的同时,矮壮素、甲哌啶也可能同时被吸附了,所以不再考虑Carb,因此采用100 mg C18+100 mg PSA+300 mg MgSO4、100 mg C18+300 mg MgSO4、100 mg PSA+300 mg MgSO43种净化剂一定比例的组合,所得的回收率结果(图4)。结果发现,对于矮壮素、甲哌啶、抑芽丹等几种极易被吸附的植调剂,发现用PSA净化的回收率能满足要求,优于其他组的搭配的净化剂。在这种方法下,16种植调剂的平均回收率在71.5%~99.3%,满足残留检测要求。
2.4 线性范围和检出限 在优化的条件下进行了方法学的考察。用甲醇配制16种植调剂的混合标准溶液,质量浓度分别为0.01、0.02、0.05、0.1、0.2、0.5mg/L,进样2μL,以MRM定量离子的色谱峰面积(Y)对分析物质量浓度(X, mg/L)建立标准曲线,相关系数(R2)在0.986 6~0.999 7之间,表明各化合物在相应的浓度范围内呈良好的线性关系。以3倍信噪比(S/N)确定检出限(LOD),得到16种农药的LOD为0.03~1.0 μg/kg。列出了16种植调剂的保留时间、线性方程、相关系数及检出限(表2)。按照保留时间先后分别列出16种植调剂各自的色谱图和质谱图(图5)。
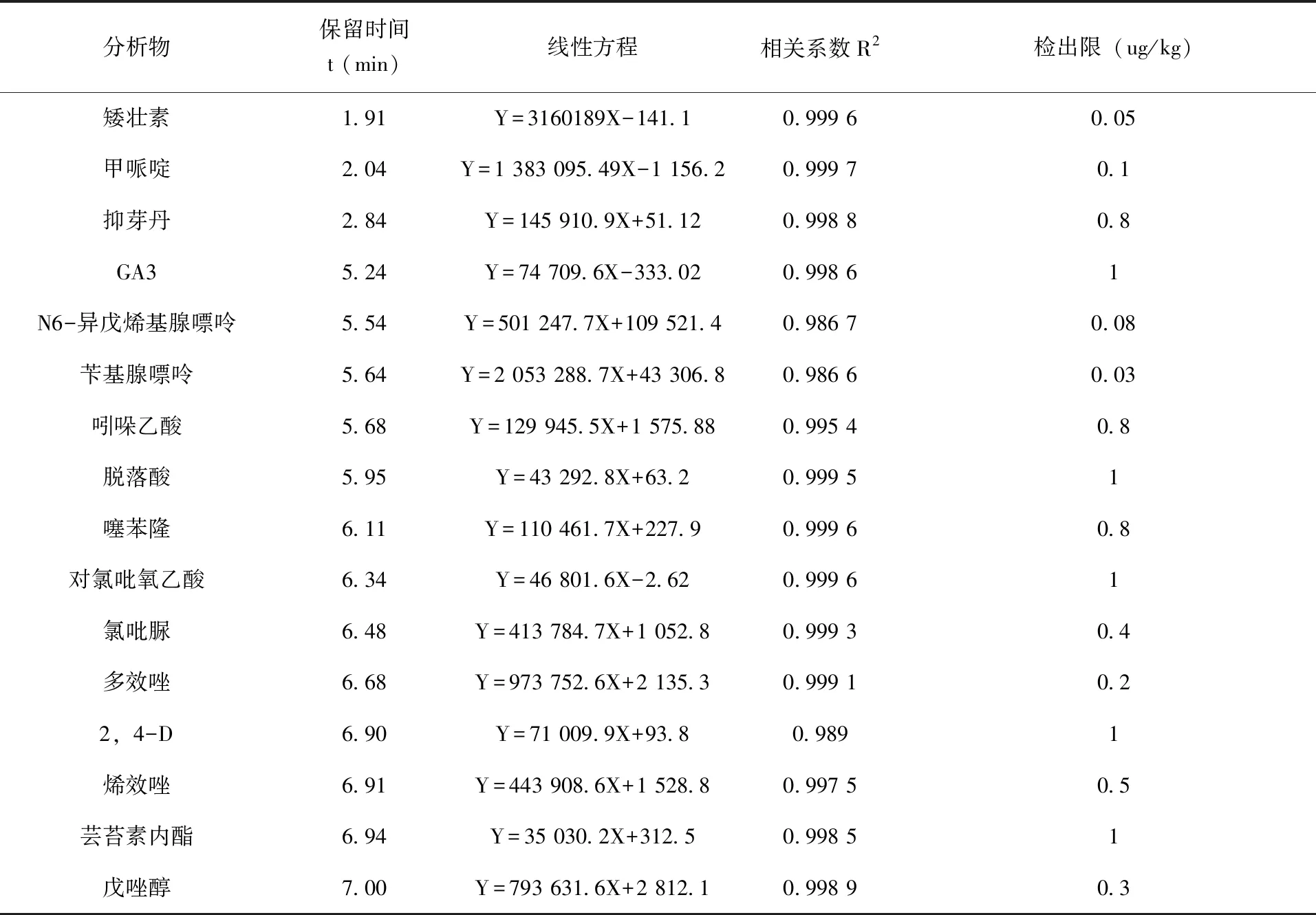
表2 16种植调剂的保留时间、线性方程、相关系数、检出限
2.5 回收率、精密度和定量限 选用不含目标化合物的两种葡萄空白样品阳光玫瑰(绿皮葡萄)和巨玫瑰(紫皮葡萄),通过添加回收试验,考察了方法的回收率和精密度(n=5)。16种植调剂的添加浓度水平为0.05、0.10、0.50mg/kg,结果表明,16种植调剂的平均加标回收率范围为71%~114.5%,RSD在0.9%~17.8%之间,方法的回收率和精密度均符合残留分析要求。

表3 阳光玫瑰和巨玫瑰葡萄中16种植调剂的回收率、精密度和定量限

续表
3 小结
本文通过对提取溶剂、净化剂和质谱条件的优化,建立了超高效液相色谱-串联质谱法同时测定葡萄中16种植物生长调节剂残留量的方法。该方法简便快捷、灵敏可靠,线性、准确度和精密度较好,同时提高了实验室的检测速度,为葡萄中16种植调剂的多残留检测、风险评估等研究提供了一种高效、可靠的分析手段。
