白粉菌诱导簇毛麦叶片酵母双杂交文库构建及CMPG1-V候选互作蛋白筛选
2020-07-28张恒陈怡名张旭牛影赵佳吴承云郝永利孙丽王海燕肖进王秀娥
张恒,陈怡名,张旭,牛影,赵佳,吴承云,郝永利,孙丽,王海燕,肖进,王秀娥
(南京农业大学细胞遗传研究所/作物遗传与种质创新国家重点实验室/江苏省现代作物生产协同创新中心,江苏 南京 210095)
小麦白粉病是由禾本科布氏白粉菌小麦专化型(Blumeriagraminisf.sp.tritici,Bgt)引起的真菌性病害,威胁小麦安全生产[1]。小麦白粉病在我国东北、华北、西北麦区普遍发生,且日趋严重[2]。目前对小麦白粉病的防治主要依赖三唑酮类人工合成的化学药剂和硫磺粉类等天然化学药剂[3]。大量农药的使用会引起环境污染、危害健康和引发食品安全等一系列问题[4]。因此,发掘抗白粉病新抗源、克隆抗性相关基因并解析其作用机制,对于小麦抗性育种具有重要指导意义。
簇毛麦(Haynaldiavillosa,又名Dasypyrumvillosum)是小麦近缘物种,为小麦族簇毛麦属的一年生或多年生二倍体异花授粉植物[5]。已有研究表明,簇毛麦具有对条锈病、叶锈病、秆锈病、白粉病和黄花叶病毒病等多种小麦病害的抗性,同时又具有分蘖能力强、生长旺盛、籽粒蛋白含量高以及耐旱耐寒耐盐等特性,是小麦遗传改良的优异三级基因库[6]。Chen等[7]发现簇毛麦及携带簇毛麦6VS染色体臂的易位系对小麦白粉病菌的所有生理小种表现免疫或高抗,该抗病易位系已经在我国小麦抗病育种中被广泛利用。为解析簇毛麦高抗白粉病的分子机制,迄今在簇毛麦中已经克隆了多个小麦白粉病抗性相关基因,包括NLR1-V[8-9]、STPK-V[10]、LecRK-V[11]、PDI-V[12]和CMPG1-V[13]等,转基因试验证明,它们均可不同程度提高普通小麦对白粉病的抗性。进一步深入解析簇毛麦广谱抗白粉病机制对于小麦白粉病抗性改良具有重要意义。
植物受到病原菌侵染后,会启动多个蛋白的表达变化,它们协同作用启动植物的抗性通路。实验室前期研究中,利用大麦基因芯片技术从高抗白粉病的簇毛麦中克隆了1个E3泛素连接酶基因CMPG1-V(GenBank:HM241654.1)。受白粉菌诱导后,CMPG1-V在簇毛麦叶片和茎中快速上调表达,CMPG1-V转基因小麦表现苗期及成株期对白粉病的广谱抗性,且其抗性的发挥可能和水杨酸(SA)、脱落酸(ABA)及H2O2信号通路相关。蛋白结构分析发现,CMPG1-V为含有1个U-Box结构域和1个ARM结构域的PUB蛋白,其泛素化活性依赖于U-Box结构,推测其通过泛素化其互作蛋白在小麦抗白粉病中发挥作用[13]。E3泛素连接酶是植物泛素化途径的关键元件,通过与其他被其泛素化的蛋白互作发挥生物学功能,因此,发现CMPG1-V的互作蛋白是解析其抗性分子通路的关键。目前研究植物蛋白互作的方法主要有免疫共沉淀(Co-IP)、酵母双杂交(Y2H)、双分子荧光互补(BIFC)、Pull-down、蛋白质芯片技术(protein chip)和邻近生物素识别技术(BioID)等[14]。其中酵母双杂交技术是Fields等[15]利用酵母转录因子GAL4的特性创造出的一种检测蛋白与蛋白之间互作的技术,该方法不但可以检测已知蛋白之间的互作关系,还可以筛选与已知蛋白互作的未知蛋白,是高通量检测蛋白互作的重要技术。本研究构建了簇毛麦叶片受白粉菌诱导后不同时间段的cDNA文库,并以CMPG1-V为诱饵,筛选其可能的互作蛋白,旨在为解析CMPG1-V的抗性机制奠定基础。
1 材料与方法
1.1 植物材料及处理方法
簇毛麦品系91C43(Haynaldiavillosa,2n=2x=14,基因组VV)引自英国剑桥植物园。簇毛麦种子先置于含水的培养皿中,25 ℃黑暗培养22~24 h,直至种子吸胀至露白;然后将露白的种子有序转移至铺有湿润滤纸的直径为15 cm的培养皿中,待种子发芽并生长至根长和叶长约为2~3 cm时,再将小苗转移至百孔盒中用霍格兰营养液培养[16],每2 d更换1次营养液;发芽及培养条件为昼/夜温度24 ℃/16 ℃,相对湿度70%,昼/夜光周期14 h/10 h;待生长至2叶1心期时,将在感白粉病品种‘苏麦3号’上繁殖的新鲜白粉菌孢子(生理小种E26,由中国农业科学院植物保护研究所周益林研究员惠赠)均匀抖落在簇毛麦叶片表面进行人工接种,分别在接种后0、1、2、4、6、12、24和48 h剪取叶片,迅速置于液氮中冷冻,-80 ℃保存备用。
烟草(Nicotianatabacum)品种‘NC89’保存于本实验室。种子直播于直径为5 cm的花盆中,生长条件为昼/夜温度24 ℃/22 ℃,相对湿度70%,昼/夜光周期 12 h/12 h。待烟草生长至2叶期(约10~12 d)时,将其单独移栽到蛭石与营养土质量比为1∶1的混合土中,相同生长条件培养至5~6叶期(30 d左右),挑选生长状态良好的植株用于双分子荧光互补实验。
1.2 菌株和载体
酵母菌株Y187和Y2HGold,载体pGADT7-Rec(SmaI线性化载体)、pGADT7-T,pGADT7、pGBKT7-53、pGBKT7-Lam和pGBKT7均购自TaKaRa公司。酵母菌株AH109保存于本实验室。农杆菌菌株GV3101购于北京鼎国昌盛生物技术有限公司。
1.3 主要试剂
Trizol Reagent,苯酚、氯仿、异戊醇混合液(体积比25∶24∶1)购自 Invitrogen公司;TaqDNA Polymerase购自诺唯赞公司;DL 2000 DNA Ladder、琼脂糖凝胶 DNA 回收试剂盒、质粒小提试剂盒购自天根生化科技有限公司;Yeast Media Set 2 Plus试剂盒(Cat.No.630495)、MatchmakerTMLibrary Construction & Screening Kits(Cat.No.630445)、NucleoSpin RNAⅡ Kits(Cat.Nos.740955.20)、NucleoTrap mRNA Kits(Cat.Nos.740655),以及培养基YPDA、SD/-Trp、SD/-Leu、SD/-Leu/-Trp(DDO)、SD/-Ade/-His/-Leu/-Trp(QDO)、Aureobasidin A、X-a-Gal均购自TaKaRa公司。乙酰丁香酮、MES、氯化镁(MgCl2)、AgarB、醋酸锂(LiAc)、YNB、大豆蛋白胨等各类基本培养基所需试剂均购于北京鼎国昌盛生物技术有限公司。
1.4 总RNA提取和mRNA分离
参照Trizol Reagent试剂说明书,提取受白粉菌诱导后0、1、2、4、6、12、24和48 h的簇毛麦叶片总RNA;分别检测所有RNA样品的质量和浓度,并按等浓度混合所有样品;使用NucleoSpin RNAⅡ 试剂盒(Cat.Nos.740955.20)对RNA进行纯化,并使用NucleoTrap mRNA 试剂盒(Cat.Nos.740655)分离总RNA中的mRNA,利用琼脂糖凝胶检测mRNA质量。
1.5 酵母双杂交cDNA文库的构建
参照Make Your Own Mate & PlateTMLibrary System(Cat.No.630490)说明书,首先使用SMART技术得到纯化的mRNA并生成双链 cDNA(ds cDNA),然后将ds cDNA与已经线性化的pGADT7-Rec载体共转化入酵母菌株Y187,并在SD/-Leu培养基上培养4~5 d,最后将100个培养皿中的所有酵母克隆收集到液体培养基中,即为酵母双杂交文库。将所得到的酵母文库分装并于-80 ℃保存备用。
1.6 库容量及重组率和插入片段长度的鉴定
从文库中取出100 μL酵母菌液,分别稀释101、102、103、104、105倍之后涂布于SD/-Leu平板,28 ℃ 培养2~3 d后对平板上生长的酵母克隆进行计数。文库滴度(CFU·mL-1)=平板克隆数×稀释倍数/涂板体积。从SD/-Leu平板上随机挑取24个酵母单克隆,使用SD/-Leu液体培养基培养24~48 h后,利用PCR技术对其插入片段进行鉴定,PCR引物分别为pGADT7 Forward(5′-TTCGATGATGAAGATACCCCACCA-3′)和pGADT7 Reverse(5′-GTGAACTTGCGGGGTTTTTCAGTA-3′)。PCR反应条件:95 ℃ 预变性 5 min;95 ℃变性 30 s,56 ℃ 复性 30 s,72 ℃ 延伸 3 min,32个循环;72 ℃ 延伸 10 min;4 ℃ 保存。PCR产物使用 15 g·L-1琼脂糖凝胶电泳进行检测,计算重组率及插入片段长度。
1.7 诱饵载体的构建及自激活检测
根据CMPG1-V基因CDS及pGBKT7载体酶切位点,设计特异引物(BD-CMPG1-V Forward:GCCATGGAGGCCGAATTCATGGTCACGCCGCTGCCCC,BD-CMPG1-V Reverse:TGCAGGTCGACGGATCCTCAGAAG-GGTCTCTTG),利用同源重组的方法将CMPG1-V插入到pGBKT7载体的EcoRⅠ和BamHⅠ2个酶切位点之间,构建诱饵载体pGBKT7-CMPG1-V。将pGBKT7和pGBKT7-CMPG1-V载体转入酵母菌株Y2HGold,分别挑取阳性单克隆涂布于SD/-Trp、SD/-Trp/X-α-Gal和SD/-Trp/X-α-Gal/AbA培养基,观察菌落是否生长或者变蓝。若菌落在SD/-Trp培养基上可正常生长,表明pGBKT7及其融合载体成功转入酵母菌株Y2HGold;若菌落在SD/-Trp/X-α-Gal培养基上生长但不变蓝,表明插入基因无自激活活性,若生长且变蓝,表明插入基因有自激活活性;当菌落在SD/-Trp/X-α-Gal培养基上生长且变蓝,在SD/-Trp/X-α-Gal/AbA培养基上生长但不变蓝,表明AbA可以抑制插入基因的自激活活性;反之,若在SD/-Trp/X-α-Gal/AbA培养基上生长且变蓝,则AbA不能抑制插入基因的自激活活性。
1.8 CMPG1-V互作蛋白的筛选及回补验证
在SD/-Trp平板上挑取转入pGBKT7-CMPG1-V载体的Y2HGold菌落,在50 mL SD/-Trp培养基中扩大培养(培养条件:250~270 r·min-1,30 ℃)至D600为0.8左右;1 000g离心弃上清液,然后用4~5 mL SD/-Trp培养基重悬菌液并移至2 L锥形瓶;向菌液中加入1 mL文库菌液和45 mL 2×YPDA液体培养基(含有50 μg·mL-1卡那霉素);30 ℃、30~50 r·min-1培养20~24 h,观察是否出现酵母合子细胞;1 000g离心弃上清液,并用50 mL 0.5×YPDA清洗锥形瓶,收集所有酵母细胞;然后用10 mL 0.5×YPDA重悬菌液,涂布于SD/-Leu/-Trp/X-α-Gal/AbA(DDO/X/A)培养基,30 ℃ 培养3~5 d。将DDO/X/A培养基上生长且变蓝的菌落挑至SD/-Ade/-His/-Leu/-Trp/X-α-Gal/AbA(QDO/X/A)培养基,进一步挑选阳性克隆。若酵母克隆在QDO/X/A培养基上继续生长且变蓝,则为真实互作;若酵母克隆无法正常生长或生长但不变蓝则可能为假阳性。
回补验证:随机挑选10个生长于QDO/X/A培养基上的阳性克隆,分别提取其pGADT7重组载体质粒;然后将以上pGADT7重组载体分别与pGBKT7-CMPG1-V载体共同转入酵母菌株AH109,涂布于DDO培养基,观察酵母是否正常生长。将生长正常的酵母转移至QDO/X培养基,观察酵母是否生长或变蓝。若生长且变蓝,则证明为真实互作;若不生长,或生长但不变蓝,则可能为假阳性。pGBKT7-53(p53)/pGADT7-T(T-antigen)和pGBKT7-Lam(Lam)/T-antigen分别为阳性对照和阴性对照。
1.9 双分子荧光互补(BiFC)试验
根据CMPG1-V和MYB25-V基因CDS及YC(YFP的C端)和YN(YFP的N端)载体酶切位点,设计特异引物(YC-CMPG1-V Forward:AAGGAAAAAAGCGGCCGCATGGTCACGCCGCTG,YC-CMPG1-V Reverse:CTAGTCTAGATCAGAAGGGTCTCTT;YN-MYB25-V Forward:AAGGAAAAAAGCGGCCGCATGGGGAGGGCG,YN-MYB25-V Reverse:CTAGTCTAGACTACACTAGTGA)。将CMPG1-V与YC载体融合构建YC-CMPG1-V载体,将MYB25-V与YN载体融合构建YN-MYB25-V载体。
BiFC试验参照Zhao等[17]的方法:将构建好的载体分别转化农杆菌菌株GV3101,等比例混合含有 YC+YN-MYB25-V或YC-CMPG1-V+YN或YC-CMPG1-V+YN-MYB25-V或YC+YN载体的菌株,5 000 r·min-1离心5 min后弃上清液,用注射缓冲液[10 mmol·L-1MES(pH5.6),0.1 mmol·L-1乙酰丁香酮,10 mmol·L-1MgCl2]重悬,使菌液的终浓度约为D600=1.5。用一次性注射器将重悬的菌液注入烟草叶片背面,注射后的烟草置于弱光环境中培养48~72 h,使用激光共聚焦显微镜(LSM780,Zeiss)观察荧光信号并拍照。观察时使用DAPI(4′,6-二脒基-2-苯基吲哚)染料对细胞核进行染色[18]。
1.10 基因表达分析
簇毛麦的培养及白粉病菌处理同1.1节。在处理后0、2、6、12、24、36、48和72 h,分别剪取叶片并迅速置于液氮中冷冻。使用Trizol Reagent提取RNA并反转为cDNA,-20 ℃保存。使用Louts PCR 480仪器根据AceQ SYBR Green(Vazyme)试剂说明,利用RT-qPCR分析受白粉菌诱导后簇毛麦MYB25-V的表达特征(特异引物:MYB25-V-Q Forward为TGGCTCAACTACCTCCGC,MYB25-V-Q Reverse为TGTTCCAGTGGTTCTTGACG)。反应条件:95 ℃ 预变性 1 min;95 ℃ 变性 10 s,60 ℃ 复性 30 s,41个循环;4 ℃ 保存。每个样品进行3次生物学重复。
从基因表达网站Triticeae Multi-omics Center wheat gene expression(http://202.194.139.32/expression/index.html),下载小麦TaMYB25基因受白粉菌诱导后表达数据[19],然后根据TPM(tags per million)和方差值绘图。
2 结果与分析
2.1 簇毛麦总RNA提取、mRNA纯化和ds cDNA的合成
簇毛麦叶片经白粉菌诱导后8个时间点的总RNA 的D260/D280=1.982,可见3条清晰的条带,分别为28S、18S和5S,未见弥散条带,表明RNA质量良好且未发生明显降解(图1-A);mRNA在300~5 000 bp呈弥散状分布(图1-B);ds cDNA长度主要分布在500~5 000 bp(图1-C),表明质量良好,可用于后续文库构建。
2.2 簇毛麦酵母双杂交文库的构建及评价
从获得的总体积为200 mL酵母双杂交文库中随机取出100 μL酵母菌液,稀释101~105倍,涂布于SD/-Leu平板培养3 d。在稀释105倍的平板上共统计到95个单克隆,其文库滴度为9.5×107CFU·mL-1,可以满足试验需求。随机挑取24个酵母单克隆,利用PCR技术对其插入片段进行鉴定。结果显示,所有克隆均能扩增出单一条带,且插入片段在250~2 000 bp,重组率为100%(图2)。表明文库质量良好,可用于后续试验。
2.3 以CMPG1-V为诱饵筛选簇毛麦cDNA文库
含有pGBKT7和pGBKT7-CMPG1-V载体的酵母菌落在SD/-Trp培养基上可以正常生长,在SD/-Trp/X-α-Gal培养基上也可以正常生长,但菌落非蓝色,在SD/-Trp/X-α-Gal/AbA培养基上则不能生长(图3-A)。表明CMPG1-V蛋白无自激活活性,可用于互作蛋白的筛选。
将含有pGBKT7-CMPG1-V载体的Y2HGold菌株和含有文库的Y187菌株共培养,20 h后可观察到酵母合子细胞(图3-B),表明筛库过程正常;24 h后将酵母细胞涂布于DDO/X/A培养基,生长3~5 d后,挑取可以在DDO/X/A培养基上生长且变蓝的酵母菌落,转移至QDO/X/A培养基培养,发现有79个酵母菌落可正常生长且为蓝色,其余酵母菌落则无法正常生长(图3-C)。表明所构建的簇毛麦cDNA文库可用来筛选CMPG1-V的互作蛋白。
2.4 部分互作蛋白的回补验证
从筛选到的79个阳性克隆中随机选择10个阳性克隆,分别提取pGADT7融合载体质粒DNA并测序,根据序列相似性注释其生物学功能,发现这些克隆涉及逆境响应、免疫调节和植物发育等(表1),其中5个cDNA(CMIN1、CMIN5、CMIN6、CMIN8、CMIN10)包含基因全长序列。利用Y2H回补试验对筛选到的互作阳性克隆进行验证。将pGADT7融合载体与pGBKT7-CMPG1-V载体共转入酵母菌株AH109,涂布于DDO培养基,生长3 d后,挑选生长良好的菌落转至QDO/X培养基上生长,所有菌落均可正常生长且为蓝色(图4)。回补验证结果表明文库筛选结果可靠,推测这些蛋白可以与CMPG1-V体外直接互作。
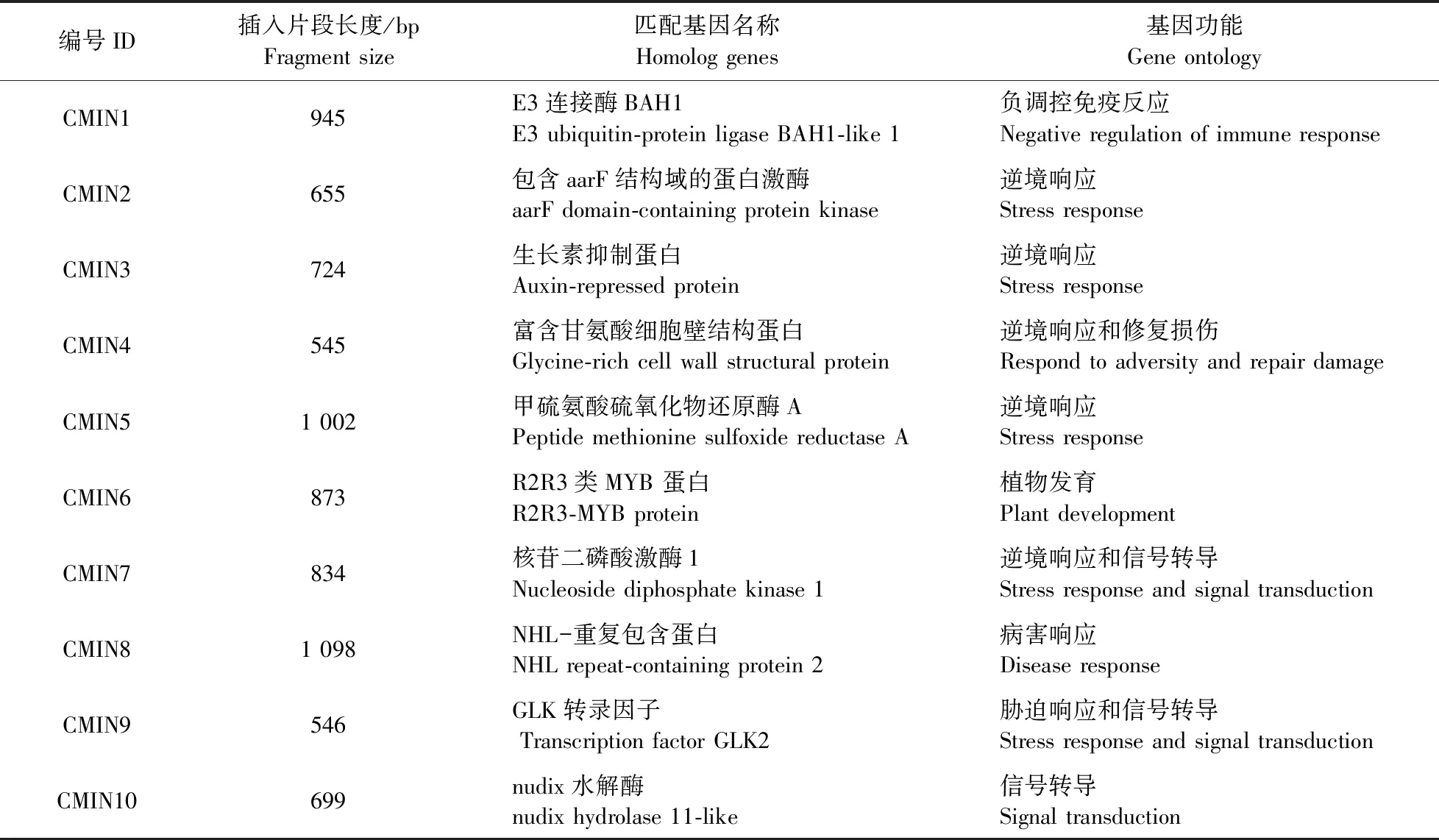
表1 10个与CPMG1-V候选互作阳性克隆的功能注释Table 1 Gene ontology of the selected 10 candidate interaction cDNA of CPMG1-V
2.5 CMPG1-V与CMIN6体内互作分析
功能注释发现CMIN6编码1个MYB转录因子。BLAST比对发现,CMIN6与小麦中的TaMYB25同源关系最近,所以将CMIN6命名为MYB25-V。将YC、YC-CMPG1-V、YN和YN-MYB25-V载体分别组合转入烟草表皮细胞,利用BiFC分析MYB25-V与CMPG1-V是否可以体内互作,发现共转化YC+YN、YC-CMPG1-V+YN或YC+YN-MYB25-V组合均不能观察到荧光信号,只有共转化YC-CMPG1-V+YN-MYB25-V时,可以在细胞核中观察到荧光信号(图5)。表明CMPG1-V与MYB25-V在烟草体内可以互作,且互作主要发生在细胞核中。
2.6 MYB25-V受白粉菌诱导表达分析
前期研究证明,CMPG1-V正向调控小麦对白粉病的抗性[13]。本研究利用公共数据库中基因表达数据,分析小麦同源基因TaMYB25受白粉菌诱导后表达特征。结果表明,TaMYB25受白粉菌诱导48 h后呈上调表达趋势,并持续至72 h(图6-A)。利用RT-qPCR分析簇毛麦叶片中MYB25-V受白粉菌诱导的表达模式,结果表明,MYB25-V在诱导后6 h开始上调表达,36 h表达量达到最高峰,并在之后的取样时间点均维持高水平表达(图6-B)。推测MYB25-V在小麦抗白粉病中也发挥作用,但CMPG1-V和MYB25-V互作与抗性的关系有待进一步研究。
3 讨论
酵母双杂交技术不但可以用来检测2个已知蛋白之间的互作关系,还可以大批量筛选某个已知蛋白的未知互作蛋白,有助于研究蛋白的功能和解析其作用机制。因此,酵母双杂交文库的构建是许多科学研究的基础工作[20]。目前,拟南芥、水稻、小麦、棉花、辣椒和玉米等作物均构建了不同处理条件下的酵母双杂交文库[21-26]。酵母文库的构建主要使用Clontech公司Mate&PlateTM文库构建系统[24-26]。该方法利用SMART技术分离纯化mRNA,并合成ds cDNA,去除了mRNA中的杂质及短片段,为文库的质量提供了保证。与传统文库构建方法相比[27-28],Mate&PlateTM文库构建系统将ds cDNA和pGADT7-Rec载体共转入酵母菌株Y187,利用酵母自身系统完成插入片段与载体的重组,省去大肠杆菌克隆、酶切和质粒提取等过程,节约建库时间,也提高了重组效率。酵母双杂交文库的筛选主要有共转化和交配2种,而使用Mate&PlateTM构建的文库是通过2种酵母的交配筛选互作蛋白,相对于共转化其筛选效率更高[29]。因此,本研究使用Mate&PlateTM系统构建簇毛麦cDNA酵母双杂交文库,这为后期文库筛选提供了方便。
cDNA文库质量主要由mRNA的完整性和文库滴度决定[23]。本研究通过Trizol提取簇毛麦的总RNA,其D260/D280为1.982,凝胶图像清晰;分离纯化的mRNA在300~5 000 bp呈弥散状分布,合成的 ds cDNA 在500~5 000 bp呈弥散状分布,获得较高丰度的ds cDNA,为构建文库奠定了基础。一般认为低丰度文库筛选要求的滴度为1×106CFU·mL-1[23],本研究构建的簇毛麦酵母双杂交文库滴度为9.5×107CFU·mL-1,可以满足低丰度筛选要求。文库平均插入片段长度为250~2 000 bp,通过对10个筛选出的cDNA测序发现,其中5个包括候选基因的全长,表明构建的簇毛麦cDNA文库质量较好。
小麦白粉病是危害小麦生产的主要病害之一。抗病基因发掘和抗性机制解析对于安全高效防治该病害有重要意义。目前,已定位80余个小麦白粉病抗性位点,并克隆了8个小麦白粉病抗性基因[1,8,9,30-35]。但以前大面积利用的Pm8等基因已经被新的白粉菌生理小种所克服[36],迫切需要发掘新的白粉病抗性基因,特别是发掘和利用具有广谱、持久抗性的新基因。U-box类E3泛素连接酶是植物中广泛存在的一类蛋白,其广泛参与植物-病原菌的互作[37]。拟南芥PUB12/PUB13、PUB22和水稻SPL11均负向调控PTI(模式识别受体触发的免疫反应)途径[38-40];而拟南芥PUB17、水稻PUB44和烟草ACRE276、ACRE74均能正向调控ETI(效应因子触发的免疫反应)途径[41-43];小麦TaPUB1正向调控小麦对盐胁迫的耐受性[44]。本实验室前期在簇毛麦中克隆了1个E3泛素连接酶基因CMPG1-V,正向调控普通小麦对白粉病的广谱抗性,该基因受白粉菌诱导快速上调表达,转基因小麦对多个白粉菌生理小种都表现出高抗[13]。本研究构建了簇毛麦cDNA酵母双杂交文库,通过文库已筛选出其互作蛋白,这为广谱抗性机制解析和新基因发掘奠定了基础。
高等植物通常拥有一大类MYB转录因子基因家族,其中许多基因被发现参与植物-病原菌的互作[45]。拟南芥中的MYB15可正向调控拟南芥对丁香假单胞菌的基础抗性[46];在普通小麦品种‘Suwon11’中沉默TaMYB4可以降低其对条锈菌CYR23的抗性[47];‘扬麦16’中过量表达TaPIMP2(编码MYB类蛋白)可以提高其对小麦根腐病的抗性[48]。部分MYB类蛋白还可与其他蛋白互作,共同调控植物与病原菌互作过程。水稻MYBS1通过抑制bsr-d1基因的表达来增加ROS的积累,限制病原菌的生长,从而增强水稻对稻瘟病的抗性[49];拟南芥MIEL1可以与MYB30互作,并导致MYB30降解,从而提高拟南芥的感病性[50];在小麦中,TuMYB46L与TuACO3的启动子结合,共同调控普通小麦对白粉病的抗性,当白粉菌入侵时,TuMYB46L基因下调表达,导致TuACO3上调表达,并促进乙烯(ET)的合成以抵抗白粉菌的侵染[45]。本研究以CMPG1-V为诱饵筛选簇毛麦酵母双杂交文库,筛选到1个MYB类蛋白MYB25-V,回补验证及BiFC试验进一步验证MYB25-V与CMPG1-V直接互作。MYB25-V与其在小麦中的同源基因TaMYB25受白粉菌诱导后均上调表达,推测MYB25-V在小麦抗白粉病中发挥作用。已有研究表明,CMPG1-V正向调控小麦白粉病抗性[13],二者互作是否及如何调控白粉病抗性有待进一步研究。
