液相色谱-四极杆/飞行时间质谱法快速筛查测定药物中芬太尼类物质
2020-07-27邓慧芬张建莹卞学海赵祥龙
邓慧芬, 张建莹*, 卞学海, 罗 耀, 赵祥龙
(1. 深圳海关食品检验检疫技术中心, 广东 深圳 518045; 2. AB SCIEX亚太应用中心, 上海 201206)
芬太尼,是一种强效的麻醉性阿片类镇痛药,于1960年由比利时药理学家和药剂师保罗·杨森研制出[1],并作为静脉麻醉剂广泛应用于临床手术麻醉中,其主要机制与吗啡相似。由于芬太尼的特殊药理性质,以及灵活的通过静脉、皮肤和黏膜给药方式,芬太尼已成为治疗疼痛最重要的阿片类药物之一。随着对芬太尼药物研究的不断深入,以其为先导化合物,通过结构改造和构效关系的评价,设计、合成、筛选到了一系列药效更强的芬太尼类衍生物,它们大多通过以下一个或多个方式得到(见图1): 1.使用其他酰基代替芬太尼的丙酰基(如乙酰芬太尼、丁酰芬太尼、丙烯酰芬太尼、呋喃芬太尼、奥芬太尼); 2.使用其他任意基团(氢原子除外)替代苯乙基部分(如β-羟基硫代芬太尼); 3.哌啶环上存在烷基、烯基、烷氧基、酯基、醚基、羟基、卤素、卤代烷基、氨基及硝基等取代基;4.使用任何取代或未取代的单环芳香基团替代与氮原子直接相连的苯基。
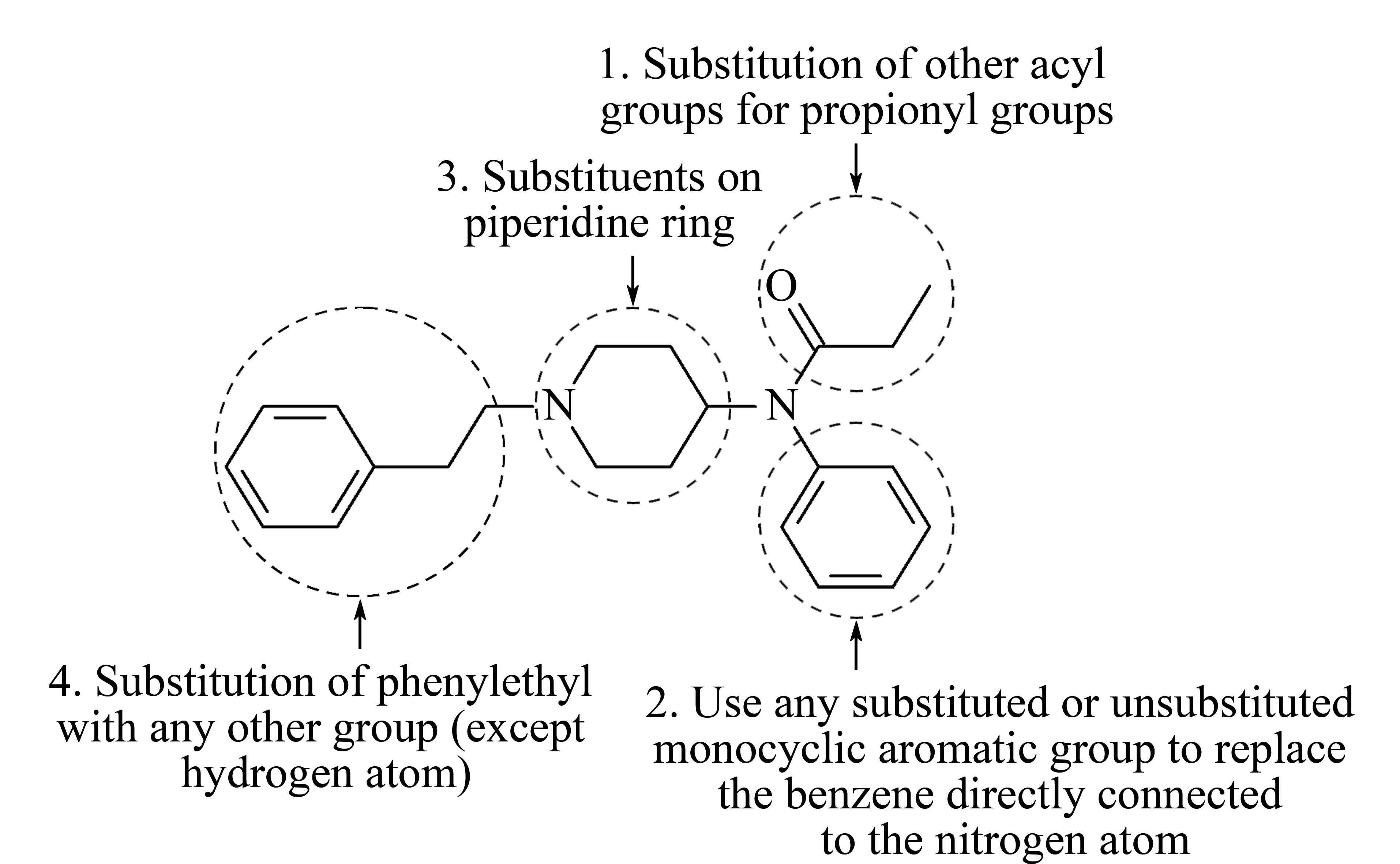
图 1 芬太尼及芬太尼类物质的结构式Fig. 1 Chemical structures of fentanyl and fentanyl and its analogues
作为药品研制出来的芬太尼却在一些国家被一些人作为毒品使用。芬太尼类物质药效较强,如芬太尼衍生物卡芬太尼,其药效和毒性是海洛因的5 000倍,约2 mg剂量就足以使一个成年人死亡[1]。美国疾病预防控制中心称,2016年美国超过2万起死亡由芬太尼类物质吸食过量导致。对此,2019年5月1日起,我国将芬太尼类物质全面列入《非药用类麻醉药品和精神药品管制品种增补目录》,开发该类药物的检测方法已成为监管部门迫切需要。
目前,国内芬太尼类物质的检测方法主要有酶联免疫吸附(ELISA)法、气相色谱-质谱(GC-MS)法、高效液相色谱(HPLC)法[2-4]和液相色谱-串联质谱(LC-MS/MS)法[5-12],主要针对芬太尼、瑞芬太尼、舒芬太尼等单一药物进行分析,方法涉及的基质包括血液、乳汁、尿液、唾液等生物基质。我国司法鉴定技术规范(SF/Z JD0107005-2016)[10]采用LC-MS/MS法测定血液、尿液中芬太尼和舒芬太尼的含量。联合国毒品与犯罪问题办公室推荐方法[1]中,GC-MS方法只涉及4种芬太尼类物质的检测,液相色谱-高分辨质谱(LC-HRMS)高分辨方法中也只涉及10多种芬太尼类物质的筛查检测。Craig等[13]采用LC-MS/MS法测定干血中11种芬太尼类物质,定量限均为0.10 ng/mL。Melissa等[14]采用LC-MS/MS法测定了血液中18种芬太尼类物质。但较少有针对广泛的药物和毒品疑似物中芬太尼类物质的高通量筛查检测和确证方法报道,这也导致了处方芬太尼类药物的过量流行和非法芬太尼类药物的出现。单级质谱和三重四极杆质谱的低分辨率、分析速度和扫描模式决定了它们无法满足痕量药物的高通量筛查检测的要求。随着高分辨质谱技术的发展,近年来Q-TOF/MS和Orbitrap MS凭借其在质量精度、全质量数据采集、数据可溯源性和数据库检索等方面的优势,以及质谱内部件的极大改进(如飞行时间质谱的飞行管由原来的V型设计成N型),能很好地弥补先前定量不足的缺陷,已广泛应用于药物筛查检测领域[6-13]。本方法通过75%(v/v,下同)乙腈水溶液提取固体和液体药物中的芬太尼类物质,采用HLB固相萃取柱净化,QTOF的信息依赖性采集(Information dependent acquisition, IDA)扫描模式获取所有化合物的一级质谱及二级质谱信息,建立一级质谱精确质量数据库和二级质谱图库,结合色谱保留时间信息,建立了液相色谱-四极杆/飞行时间质谱(LC-QTOF-MS)筛查检测药物中27种芬太尼类物质(包括芬太尼及26种类似物或代谢物)的方法。本方法的建立将有助于在监管范围、检测技术和时效性等多方面提升芬太尼类药物的安全监管水平。
1 实验部分
1.1 仪器与试剂
液相色谱-四极杆/飞行时间质谱仪(SCIEX X500R QTOF,美国AB SCIEX公司);均质仪(美国Omni公司);涡旋振荡仪(Rax top,德国Heidolph公司);高速冷冻离心机(3K18,德国Sigma公司);高纯水发生器(Milli-Q Integral 10+,美国Millipore公司); HLB固相萃取柱(60 mg, 3 mL, Waters公司); 0.22 μm有机滤膜(上海安谱公司)。
甲醇、乙腈、甲酸、乙酸铵均为色谱纯(德国Merck公司),其他试剂均为分析纯,水为Milli-Q超纯水。27种化合物标准溶液(各化合物的质量浓度均为100 mg/L,美国Cerilliant公司)。使用乙腈稀释为1.00 mg/L和100 μg/L两种浓度的混合标准工作溶液(-18 ℃避光存放)。采用空白样品(根据待测样品的性质选择维生素C压片、头痛粉、止咳水、透皮贴剂样品为空白样品)基质液稀释混合标准工作液,配制成质量浓度为5.00、10.0、25.0、50.0和100 μg/L的系列标准溶液。
1.2 样品前处理
固体、粉末样品取适量研细成粉末,搅拌均匀。液体样品称取前要摇晃均匀。透皮贴剂样品揭去防粘层,剪碎。
称取试样1.00 g,加入5 mL 75%乙腈水溶液,涡旋溶解,再加入5 mL乙腈,涡旋30 s,超声提取15 min,涡旋混合1 min, 9 500 r/min离心5 min,吸取全部上清液于40 ℃水浴中氮吹至低于3 mL。吸取浓缩液于HLB固相萃取小柱(使用前用5 mL乙腈活化)中,待上清液全部流出小柱,再加入4 mL乙腈洗脱。收集所有流出液,于40 ℃水浴中氮吹至低于0.5 mL,加入0.5 mL水,再用乙腈定容为1.00 mL,涡旋振荡30 s, 9 500 r/min离心5 min。取上清液,过0.22 μm有机滤膜,LC-QTOF-MS测定。
1.3 色谱条件
色谱柱:Eclipse Plus Phenyl-Hexyl (150 mm×2.1 mm, 3.5 μm, 美国Agilent公司)。流动相A: 0.1%(v/v,下同)甲酸水;流动相B: 乙腈/甲醇(1∶1,含0.1%甲酸,v/v,下同)。梯度洗脱程序:0~2.0 min, 5%B; 2.0~5.0 min, 5%B~50%B; 5.0~16.0 min, 50%B; 16.0~16.1 min, 50%B~5%B; 16.1~20.0 min, 5%B。流速:0.3 mL/min。进样量:10 μL。
1.4 质谱条件
离子源和气体参数:加热电喷雾离子(HESI)源;喷雾电压5 500 V;毛细管温度325 ℃;加热器温度550 ℃;雾化气3.79 Pa;辅助气4.14 Pa;气帘气35 u;碰撞气7 u;扫描模式:IDA;正离子采集模式。TOF-MS参数:采集范围为m/z200~450,去簇电压(DP) 80 V,碰撞能(CE) 10 V,累积时间0.16 s。TOF-MS/MS参数:采集范围为m/z80~450,累积时间0.06 s,电荷数1;质谱扫描参数见表1。
1.5 质谱数据库的建立
1.5.1分辨率
高分辨质谱在合适的分辨率下可以将目标化合物与基质中的同质异位素干扰物实现基线分离,从而得到更为准确的定性结果。基于X500R QTOF-MS可采集到未知物的高分辨率质谱一级(≥35 000)和二级数据,且质谱精确度≤5×10-6(5 ppm),具有实现目标物与干扰物质完全基线分离、有效地去除基质干扰、提取离子色谱图清晰完整、可准确定性和定量等优点,本实验一级质谱和二级质谱均采用≥35 000的分辨率,得到的谱图既有母离子的精确质量数,又有二级质谱全扫描信息,完全满足定性和定量要求。图2为采用IDA扫描方式得到的芬太尼标准溶液的全扫描色谱图和TOF-MS、TOF-MS/MS质谱图。
1.5.2一级质谱的精确质量数据库
在优化的LC-MS条件下进样,对27种化合物先进行一级质谱全扫描,获得目标物的高分辨质谱信息。化合物的名称、保留时间、分子式、一级质谱理论质量数、一级质谱精确质量数、质量数偏差、2个二级碎片离子的精确质量数等信息见表1。
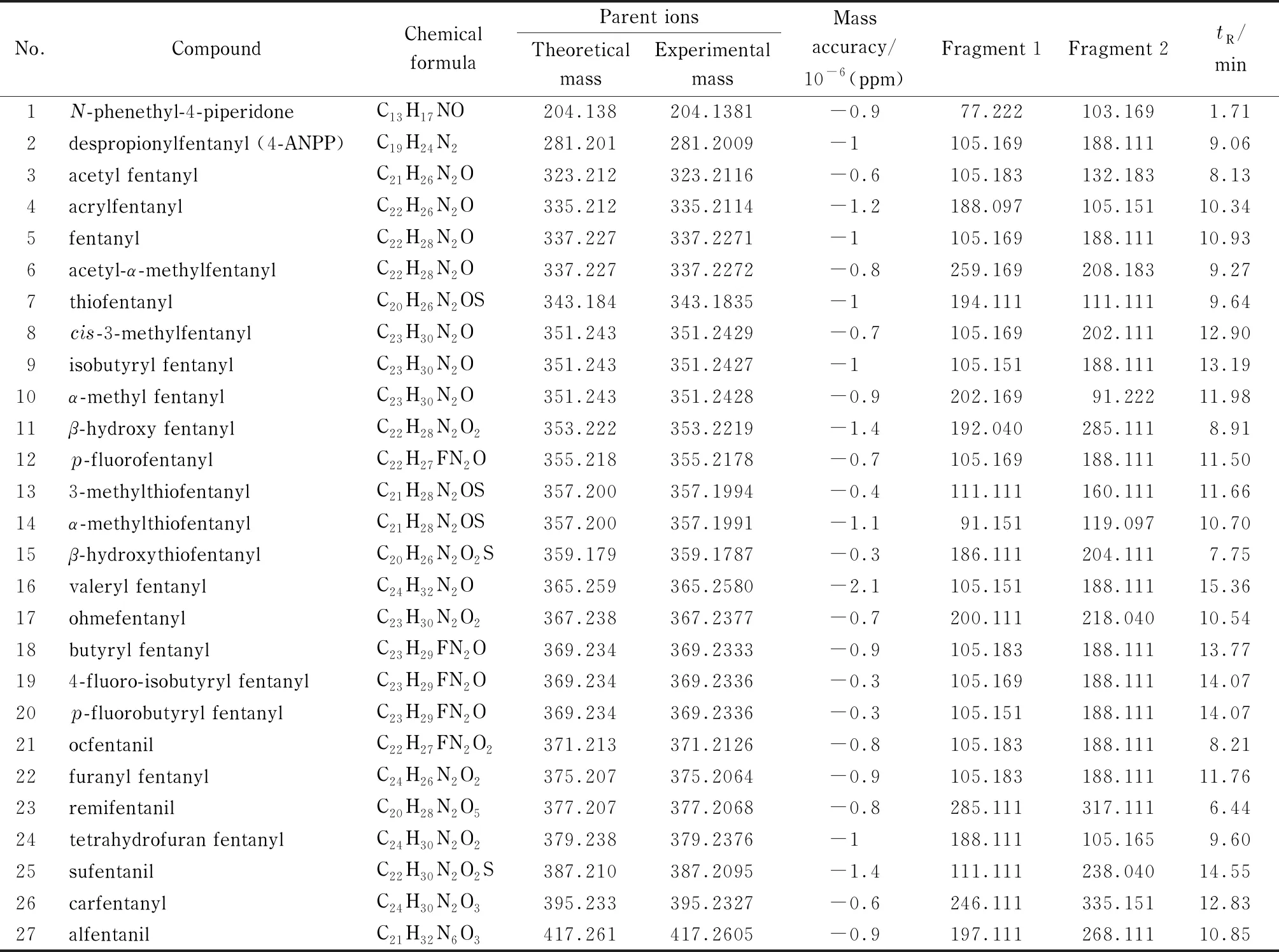
表 1 27种化合物的质谱信息和保留时间
1.5.3MS/MS二级谱库
聊了一阵子,话头转到西瓜上来了,她说:“你种的西瓜真好,甜得很。”剖了瓜,自己拿一瓣,还拿一瓣给我。我咬了两口,见上面有瓜籽,就用小指把它抠出来,瓜籽抠出来了,沾在我的指尖,我的脸也像瓜瓤子,通红通红的。
根据欧盟《食品饲料中农残分析的质量控制和方法确认的指导文件》(SANTE/11813/2017)[15]中规定的高分辨质谱定性最低要求,必须一个高分辨母离子加一个高分辨子离子,且质量偏差<5 ppm。将目标物精确质量母离子在不同碰撞能量下(20、35和50 eV)进行测定,采集并叠加二级谱图库所有的子离子信息,建立化合物的二级信息谱库。
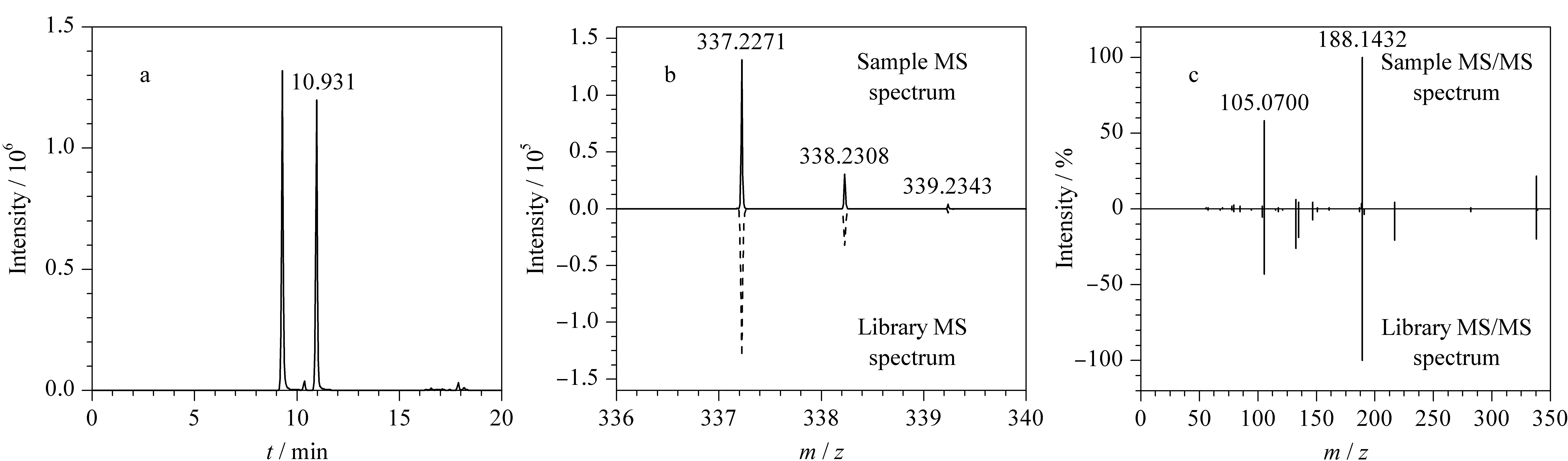
图 2 芬太尼的(a)全扫描色谱图、(b)一级质谱图和(c)二级质谱图Fig. 2 (a) Total ion chromatogram, (b) TOF-MS spectra and (c) TOF-MS/MS spectra of fentanyl
2 结果与讨论
2.1 色谱条件的选择
2.1.1色谱柱
由于芬太尼类物质具有苯基、烷基和酰胺等基团,化学性质差异较大,同时有部分同分异构体,因此本方法优先考虑使用苯基-己基(Phenyl-Hexyl)色谱柱;当使用含甲醇流动相时,其对极性/胺类化合物和芳香族化合物有显著的选择性。结果表明,在使用0.1%甲酸水-乙腈/甲醇(1∶1,含0.1%甲酸)为流动相时,27种芬太尼类物质在苯基-己基色谱柱中均得到较好的分离和保留,其中包括两类4种同分异构体(4-氟异丁酰芬太尼和4-氟丁酰芬太尼,丁酰芬太尼和对氟芬太尼)。
2.1.2流动相
考察了乙腈-水、甲醇-水和乙腈/甲醇(1∶1)-水(水相和有机相中均分别添加0.1%甲酸、5 mmol/L乙酸铵(含0.01%甲酸,pH约为4.5)和5 mmol/L乙酸铵(含0.1%甲酸,pH约为3.2))流动相条件下的色谱分离效果。在不同流动相条件下目标化合物的分离度存在差异,且质谱响应强弱不同。在甲醇-水流动相体系中,大多数化合物响应较强,分离度相对较好,但个别化合物峰形稍差。在乙腈-水流动相条件下,较多化合物峰形较好,但4种同分异构体的色谱峰分离度较差。乙腈/甲醇(1∶1)混合溶液-水流动相的优点是同分异构体分离更好,能同时兼顾峰形和分离度,这可能是由乙腈和甲醇的洗脱能力以及黏度差异导致的。在流动相中加入少量甲酸(0.1%)有利于提高分析物的离子化效率并改善色谱峰峰形,提高质谱响应强度。结果表明,以0.1%甲酸水-乙腈/甲醇(1∶1,含0.1%甲酸)为流动相比添加乙酸铵的流动相体系获得更优的色谱峰形及质谱响应。在优化的色谱条件下,27种芬太尼类物质的保留时间见表1,提取离子色谱图见图3。
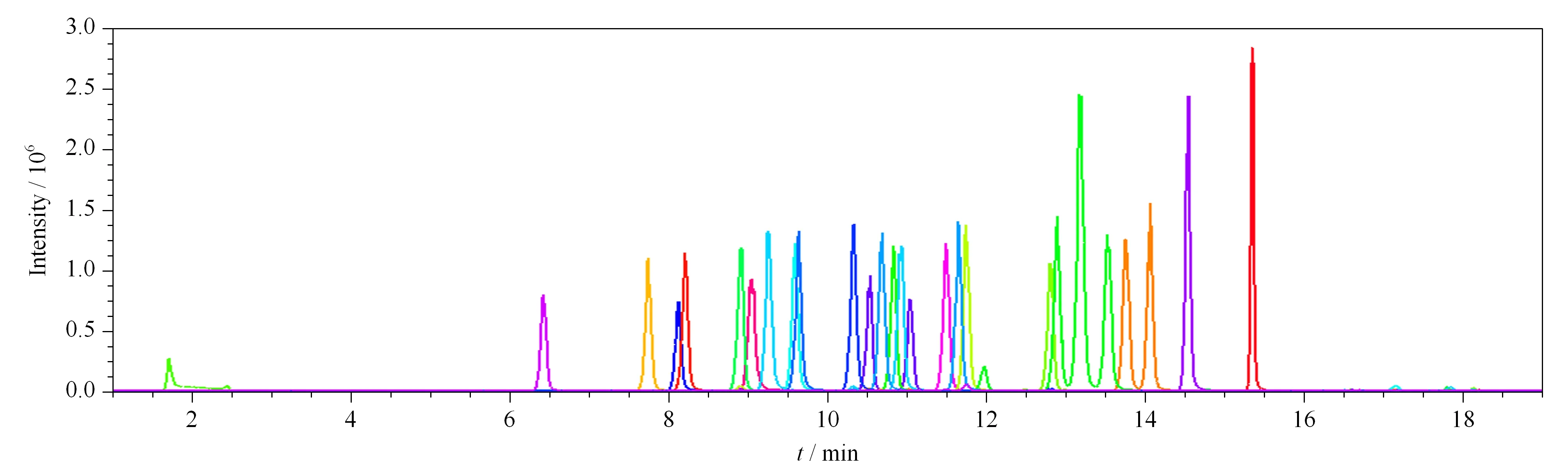
图 3 27种化合物混合标准溶液(10.0 μg/L)的提取离子色谱图Fig. 3 Extracted ion chromatogram of the 27 compound standard solution (10.0 μg/L)
2.2 样品前处理方法的优化
2.2.1提取溶剂
根据溶剂性质、目标化合物性质、样品基质特点和检测灵敏度的要求及有机溶剂含量高更有利于样液浓缩的需要,选取乙腈-水溶液作为提取溶剂。同时考察了50%乙腈水溶液、75%乙腈水溶液、100%乙腈对固体、液体药物样品中27种目标化合物的提取效果。结果表明,加入适量水,可增加固体药物的溶解,提高目标化合物的提取效率;但加入过多的水分又不利于样液的浓缩。综合考虑,选取5 mL的75%乙腈水溶液+5 mL乙腈为提取溶剂。
2.2.2净化柱
2.2.3洗脱液用量
分别比较了HLB固相萃取柱洗脱液使用2、3、4、5 mL乙腈对27种目标化合物的回收率的影响。结果表明,乙腈洗脱量为4 mL时,27种目标化合物的总体回收率较好,其回收率为73.3%~83.3%(见图4)。
2.3 基质效应(ME)
实验考察了止咳水、维生素C压片、透皮贴剂和头痛粉样品的基质效应,用纯溶剂和基质提取液配制标准曲线工作溶液,得到目标物的溶剂和样品基质匹配标准工作曲线方程,根据ME=Ka/Kb(其中Ka是指样品基质的校准曲线斜率,而Kb是指溶剂校准曲线的斜率)进行基质效应计算,并绘制基质效应图。由图5可知,这4种样品的ME值在98.0%~130%之间,说明基质效应较低。这4种基质干扰较小,样品可直接采用50%乙腈水溶液配制标准工作曲线溶液,但因其他复杂样品的基质效应未考察,因此在方法考察中统一使用基质匹配曲线。
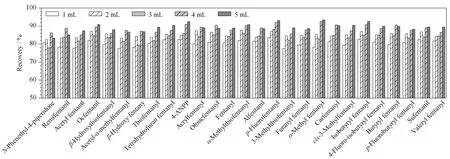
图 4 洗脱液体积对化合物回收率的影响Fig. 4 Effects of eluent volume on recoveries
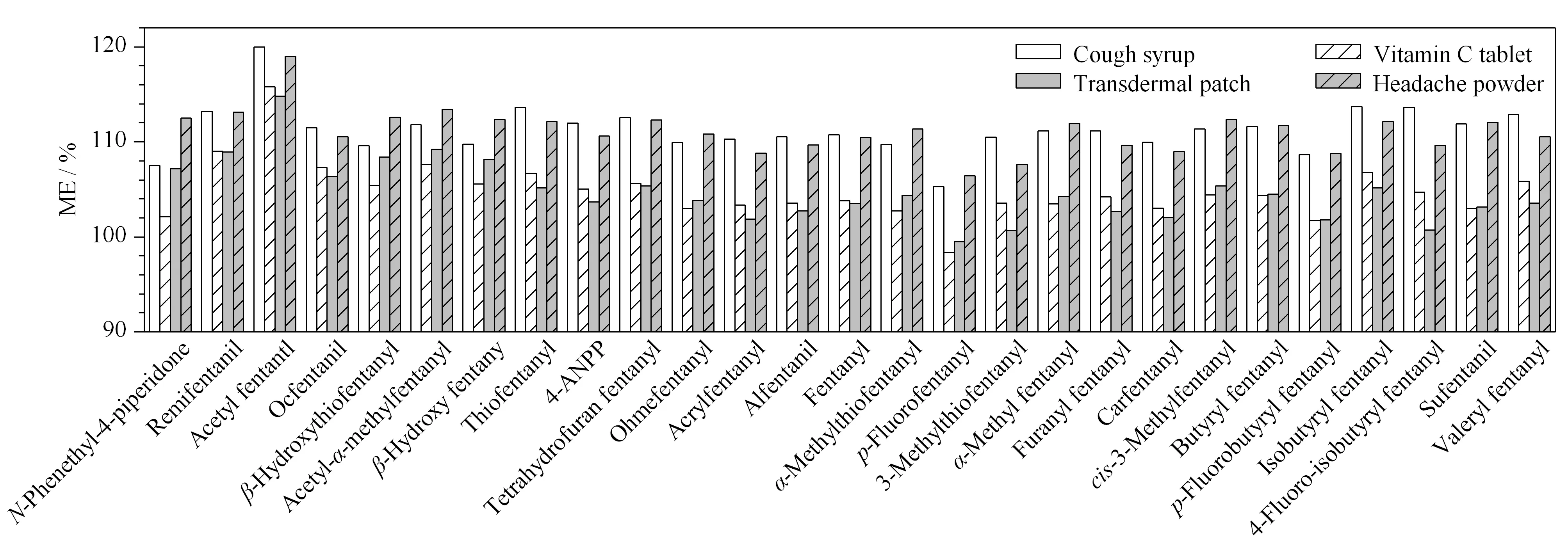
图 5 27种芬太尼类物质的基质效应Fig. 5 Matrix effects (ME) of 27 fentanyl analogs and metabolites
2.4 方法学验证
2.4.1定性方法的准确度和精密度
在维生素C压片、头痛粉、止咳水、透皮贴剂空白样品中添加27种化合物标准溶液(50.0 μg/kg)作为样品进行测定。由表1可知,27种化合物的精确质量数的偏差在3.0×10-7~2.1×10-6之间,二级全扫描质谱图的匹配度在90%以上。
2.4.2方法的线性方程和定量限
本方法在快速筛查的基础上通过提取一级质谱的精确质量数进行定量从而实现同步定量分析,每个化合物采集点数均可在20个以上,保证定量结果的准确性。以峰面积对标准溶液中各被测组分的质量浓度绘制工作曲线。27种芬太尼类物质在5.00~100 μg/L范围内线性关系良好,其相关系数和回归方程见表2。
方法的定量限是基于回收率和精密度满足欧盟SANTE/11813/2017要求而进行验证的最低浓度水平。试验结果显示,27种物质的定量限均为10.0 μg/kg。芬太尼类物质为列管物质,违禁试样主要为邮寄、运输、旅客携带的未知物、非法及处方药物,这些违禁物中芬太尼类物质的含量一般都较高,10.0 μg/kg的定量限已足够检测和监管需要。

2.4.3加标回收率和精密度
分别采用空白维生素C压片、头痛粉、止咳水、透皮贴剂样品作为验证基体。各化合物的添加水平为10.0、20.0及50.0 μg/kg,每个水平做6个平行样。按本方法中测定步骤操作,外标法定量,计算每种验证基体的回收率范围和精密度。在维生素C压片中3个添加水平下的平均回收率分别为84.8%~106%、 83.7%~96.3%和82.9%~96.2%,相对标准偏差(RSD)在0.38%~7.15%之间(n=6,见表2);在头痛粉中3个添加水平下的平均回收率分别为84.8%~106%、 87.6%~100%和85.5%~100%,RSD在0.86%~8.49%之间(n=6);在止咳水中3个添加水平下的平均回收率分别为93.0%~109%、 88.7%~97.3%和86.9%~96.9%,RSD在1.12%~7.81%之间(n=6);在透皮贴剂中3个添加水平下的平均回收率分别为83.1%~106%、 89.0%~101%和87.5%~98.1%,RSD在0.69%~8.71%之间(n=6)。
3 结论
本实验利用高效液相色谱-四极杆/飞行时间质谱对固体压片、粉末、液体、透皮贴剂中27种芬太尼类物质进行筛查测定,20 min内可完成27种芬太尼类物质的高通量筛选和确认。高分辨质谱在样品分析中消除了基质干扰,结合固相萃取前处理方法,极大提高了方法的定性准确性和定量灵敏度。以精确质量数、二级质谱图和相对保留时间为基础,构建了27种芬太尼类物质筛查数据库,建立了27种芬太尼类物质的快速筛查和确证方法。该方法作为一种国内外高度关注的新型毒品芬太尼类物质的快速筛选和确证检测技术,必将在公安缉毒、刑侦及相关检测部门对芬太尼类物质监控筛查和确认中得到广泛应用,具有应用价值。
