超高效液相色谱-串联质谱法同时测定血清中12种抗癫痫药物
2020-07-27高乐虹彭方达于丽佳王朝东王玉平丁春光
代 静, 高乐虹, 彭方达, 于丽佳, 王朝东, 王玉平, 丁春光*
(1. 国家卫生健康委职业安全卫生研究中心, 北京 102308; 2. 首都医科大学宣武医院, 北京 100053)
癫痫是一种常见的、复杂的神经系统疾病,是神经元高度同步化放电所致反复痫性发作的慢性疾病[1]。临床上癫痫通常采用单一用药或联合用药的方式进行治疗,抗癫痫药物如苯妥英钠(phenytoin sodium)、卡马西平(carbamazepine)、丙戊酸钠(sodium valproate),治疗范围狭窄,毒性较强,易发生不良反应,需要定期监测血药浓度。随着医药水平的提高,出现了很多新型抗癫痫药物如拉莫三嗪(lamotrigine)、奥卡西平(oxcarbazepine)、托吡酯(topiramate)、左乙拉西坦(levetiracetam)等[2],相较于传统抗癫痫药物,它们的治疗效果更好,毒副反应低,但需长期服药。患者在服用抗癫痫药物后存在较大的个体差异,即便是经同一途径使用相同剂量药物,也会出现各不相同的治疗反应。为了确保患者用药的安全性和有效性,在临床治疗中需要开展及时、准确的治疗药物监测(therapeutic drug monitoring, TDM),定期检测患者体内的药物浓度,制定最佳的个体化给药方案[3,4]。
目前对于抗癫痫药物的检测有色谱分析法[5-12]、光谱分析法[13]、免疫分析法[14-15]、电化学分析法[16]等,其中,色谱分析法应用较为广泛。实验室中通常采用高效液相色谱法(HPLC)或高效液相色谱-串联质谱法(HPLC-MS/MS)进行血药浓度的分析。高效液相色谱法选择性强,重现性高,但容易受到内源性杂质的干扰,同时分析多种物质难度较大。高效液相色谱-串联质谱法具有优越的定性与定量功能,专属性强,灵敏度高,受内源性杂质干扰小,能够同时进行高量物质分析的特点[17]。当前文献报道的检测方法多针对传统抗癫痫药物,且方法同时检测种类较少,不能满足临床抗癫痫药物不断增加的需求。基于此,本研究建立了同时测定血清中12种抗癫痫药物的超高效液相色谱-串联质谱法(UPLC-MS/MS),方法前处理简便、快速,专属性强,灵敏度高,大大提高了检测效率,为临床上抗癫痫药物血药浓度的监测、个体化给药方案的制订、药物代谢动力学的研究提供了方法技术支持。
1 实验部分
1.1 仪器、试剂与材料
Waters Xevo TQ-S Micro超高效液相色谱-三重四极杆质谱仪(美国Waters公司); E3116R型台式高速冷冻离心机(广东安胜仪器有限公司);十万分之一天平(德国Sartorius公司); VORTEX-5型涡旋混合器(江苏海门其林贝尔仪器制造有限公司); Milli-Q型超纯水机(美国Millipore公司)。
1.2 实验条件
1.2.1标准溶液的配制
分别精密称取适量标准品,用甲醇溶解、定容,将丙戊酸钠配制成5 mg/mL的标准储备液,其余物质配制成1 mg/mL的标准储备液,于-20 ℃冰箱中保存备用。
分别移取12种标准储备液适量,用甲醇稀释,制得混合标准储备液,其中氯硝西泮质量浓度为5 μg/mL,丙戊酸钠质量浓度为250 μg/mL,其余10种物质的质量浓度为50 μg/mL,于-20 ℃冰箱中保存备用。使用前用甲醇稀释至所需浓度。
分别精密称取非那西丁、氯唑沙宗标准品,用甲醇溶解、定容,配制成1 mg/mL的内标标准储备液,于-20 ℃冰箱中保存备用。使用前用甲醇稀释,配制得10 μg/mL的内标工作液。
1.2.2血清样品的处理
量取血清样品200 μL,加入内标工作液(氯唑沙宗10 μL、非那西丁20 μL),涡旋混匀,加入800 μL乙腈,涡旋振荡60 s,静置5 min,于4 ℃以12 000 r/min离心10 min,取上清液上机检测。
1.2.3色谱条件
经过多年发展,中国已陆续建成了西气东输、陕京管道系统、川气东送、甬沪宁、兰郑长等一批长距离、大输量主干管道,2017年底中国境内在役油气管道总里程累计约13.31万千米[1],形成了“北油南运”“西油东运”“西气东输”“海气登陆”的供应格局。
色谱柱:Waters ACQUITY UPLC BEH C18柱(100 mm×2.1 mm, 1.7 μm);柱温:50 ℃;流动相:A为水(含10 mmol/L甲酸铵), B为甲醇(含10 mmol/L甲酸铵);流速:0.4 mL/min。梯度洗脱程序:0~0.5 min, 98%A; 0.5~5.5 min, 98%A~30%A; 5.5~5.7 min, 30%A; 5.7~7.0 min, 30%A~10%A; 7.0~9.0 min, 10%A~98%A。进样量:2 μL。
1.2.4质谱条件
离子源;电喷雾电离源;离子源温度:150 ℃;去溶剂化温度:350 ℃;扫描方式:多反应监测(MRM)模式,正、负离子同时扫描;液氮气体流速:650 L/h;毛细管电压:3.5 kV。12种抗癫痫药物及内标的质谱参数见表1。
2 结果与讨论
2.1 色谱条件优化
本方法采用梯度洗脱使12种待测化合物得到较好的分离,减少了血清中内源性杂质的干扰。实验分别考察了甲醇-水、乙腈-水体系作为流动相对色谱分离的影响。甲醇-水体系作为流动相时,洗脱能力更强,出峰时间适宜,因此选择甲醇-水作为流动相。
但部分待测化合物的离子化效率较低,故考虑在流动相中加入改性剂以调节流动相的离子强度,提高化合物的响应值。实验分别在水相和有机相中添加了0.1%甲酸、10 mmol/L甲酸铵、10 mmol/L乙酸铵。结果表明,当流动相中添加10 mmol/L甲酸铵时,质谱响应最好,检测灵敏度高。
同时实验还考察了不同浓度(5、10和20 mmol/L)甲酸铵溶液对色谱分离和质谱响应的影响。当甲酸铵浓度较低或较高时,对负离子电离均有抑制,当甲酸铵溶液浓度为10 mmol/L时,结果较为理想。
因而实验最终选择10 mmol/L甲酸铵水溶液和含10 mmol/L甲酸铵的甲醇溶液为流动相。
2.2 质谱条件优化
采用电喷雾电离源,分别对12种待测化合物和内标标准溶液进行一级质谱全扫描,确定各化合物的准分子离子。在正离子模式下,普瑞巴林、左乙拉西坦、加巴喷丁、卡马西平、拉考沙胺、奥卡西平、拉莫三嗪、氯硝西泮及非那西丁均出现[M+H]+峰;在负离子模式下,苯巴比妥、托吡酯、氯唑沙宗均出现[M-H]-峰。此外,由于苯妥英钠和丙戊酸钠分子结构为钠盐,在水中会水解形成苯妥英和丙戊酸,故其准分子离子峰分别为苯妥英的[M+H]+峰及丙戊酸的[M-H]-峰。将扫描得到的准分子离子作为母离子进行二级质谱扫描,优化碰撞能量和锥孔电压,优化得到的质谱参数见表1。
2.3 方法学考察
2.3.1专属性
取6个不同来源的空白血清,混合均匀,移取200 μL,不加内标,加入800 μL乙腈,按1.2.2节描述处理并上机进样测定。同时配制加入12种混合标准溶液的样品溶液,并加入非那西丁和氯唑沙宗内标溶液后同法测定,考察12种抗癫痫药物及2种内标化合物的专属性。结果表明,基质中内源性物质不干扰待测化合物的测定,12种抗癫痫药物和2种内标化合物的总离子流色谱图见图1。

图 1 12种抗癫痫药物和2种内标化合物的总离子流色谱图Fig. 1 Total ion chromatogram of the 12 antiepileptic drugs and two internal standards 1. pregabalin; 2. gabapentin; 3. levetiracetam; 4. lacosamide; 5. lamotrigine; 6. phenobarbital; 7. phenacetin (IS); 8. topiramate; 9. oxcarbazepine; 10. chlorzoxazone (IS); 11. phenytoin sodium; 12. carbamazepine; 13. clonazepam; 14. sodium valproic.
2.3.2基质效应
在体内药物分析中,基质常常对分析物的分析产生干扰,并影响分析结果的准确性,因而需要考察基质效应。分析物和内标的绝对基质效应可以用基质因子(MF)表示:MF=As/An,其中,As为空白基质溶液中待测物或内标的峰面积;An为空白溶剂中待测物或内标的峰面积。当MF=1,表明基质效应不存在;当MF>1,可能存在离子增强效应;当MF<1:可能存在离子抑制效应。进一步将分析物的基质因子除以内标的基质因子,得到经内标归一化的基质因子。
本实验使用6批来源不同的空白血清样品,提取后分别加入12种待测化合物,制备低、中、高3个水平(0.05、0.25、1.25 mg/L)的质控样品,进样并计算经内标归一化的基质因子。结果显示,12种抗癫痫药物在血清中经内标归一化的基质因子为95.60%~108.0%, RSD值≤10.6%,能够满足测定需求。
2.3.3线性关系和定量限
取950 μL空白血清样品,分别加入已配制好的系列混合标准工作液各50 μL,涡旋混匀,配制成系列质量浓度的血清样品溶液。取200 μL制备好的血清样品,按1.2.2节描述进行前处理,然后上机测定,以待测物质量浓度(X, μg/L)为横坐标,待测物和内标峰面积的比值(Y)为纵坐标进行线性回归分析,并用最小二乘法(权重系数1/X)计算得到回归方程。12种抗癫痫药物在各自线性范围内线性良好,相关系数(r2)均大于0.992,能够满足检测要求;以信噪比(S/N)为10时待测物的质量浓度为定量限,12种抗癫痫药物的定量限范围为0.140~103 μg/L,满足定量测定需求。12种抗癫痫药物的线性范围、线性方程、相关系数和定量限见表2。
2.3.4准确度和精密度
取空白血清样品,分别配制12种抗癫痫药物低、中、高3个水平的加标样品,由于实际样品一般要稀释10倍或20倍后测定,所选的低、中、高水平基本覆盖了稀释后各药物有效血药浓度范围。低、中、高每个水平制备6个样品进样测定,考察加标回收率和批内精密度,通过测定3个分析批,考察批间精密度。实验结果见表3, 12种抗癫痫药物的加标回收率为90.80%~114.0%,批内RSD值≤13.2%,批间RSD值≤14.8%。表明方法的准确度和精密度均良好,能够满足样品的定量分析要求。
2.3.5稳定性
日内稳定性:采用低和高水平(0.05 mg/L和1.25 mg/L)质控样品,预处理后置于自动进样盘(10 ℃)中,分别于0、2、4、8、12和24 h进样测定。结果表明,12种待测物含量的RSD值≤9.14%,日内稳定性良好。
短期稳定性:采用低和高水平(0.05 mg/L和1.25 mg/L)质控样品,置于-20 ℃冰箱中冷冻保存,分别于第1、7、15 d解冻后进样测定。结果表明,12种抗癫痫药物含量的RSD值≤13.6%,说明在15 d内样品能够保持稳定。

表 2 12种抗癫痫药物的线性范围、线性方程、相关系数和定量限

表 3 12种抗癫痫药物的加标回收率和相对标准偏差
冻融稳定性:采用低和高水平(0.05 mg/L和1.25 mg/L)质控样品,置于-20 ℃冰箱中冷冻保存,反复冻融3次,前处理后进样测定。结果表明,12种抗癫痫药物测得浓度的RSD值≤14.5%,在冻融3次下均能够保持稳定。
2.4 实际样品检测
应用建立的检测方法对10例服用抗癫痫药物的患者进行血药浓度分析。根据文献[4]提到的抗癫痫药物有效血药浓度范围,多数患者体内抗癫痫药物未达到有效浓度,有1位患者体内丙戊酸钠含量超出有效浓度范围(见表4)。
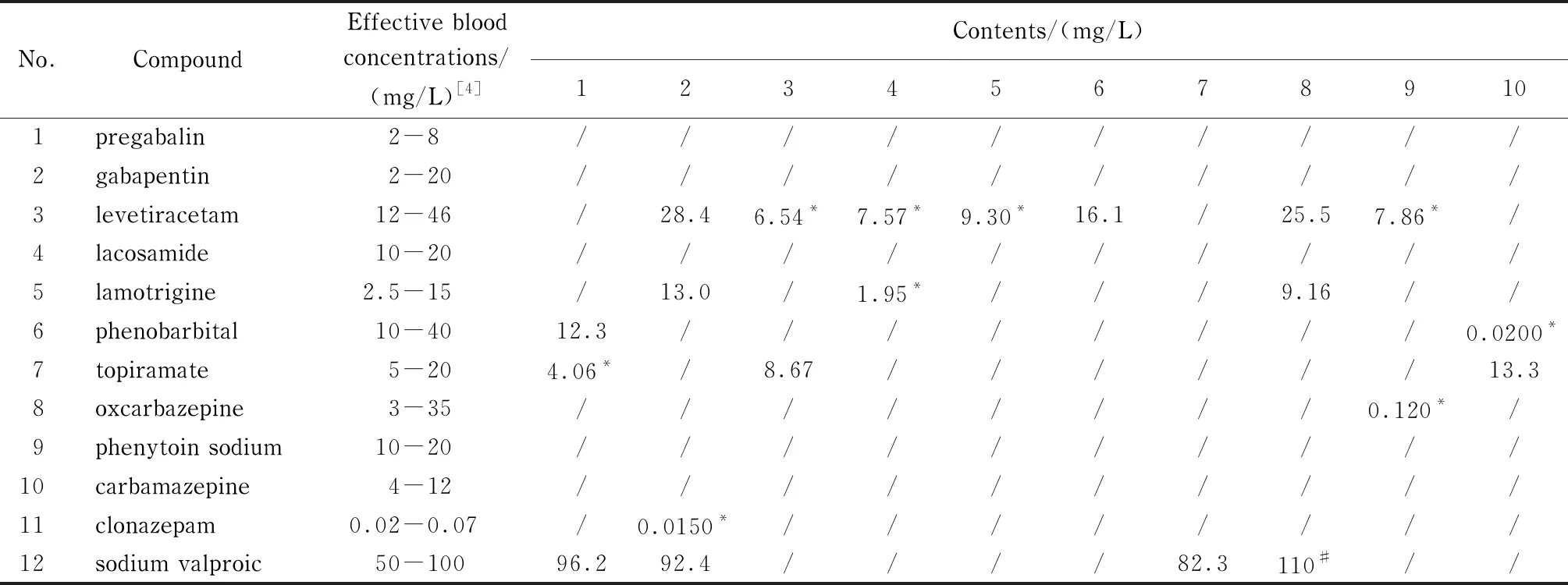
表 4 实际样品中12种抗癫痫药物的测定结果
这些检测结果可以帮助医生准确掌握患者体内的药物类型和血药浓度,从而为给患者进行精准用药提供依据。在检测过程中还发现,医生为某位患者开具了左乙拉西坦、丙戊酸钠、拉莫三嗪3种药物的处方,但这位患者体内除这3种药物外,还检出了较高含量的氯硝西泮。在治疗过程中,医生往往不能完全掌握患者曾经的用药情况,开药时可能发生用药种类重复、用药剂量过大、联合用药产生不良反应等问题,而本方法能够同时筛查12种抗癫痫药物,便于了解患者之前的用药史,从而帮助医生根据患者之前的用药情况更有针对性地进行治疗。
3 结论
本文建立了同时测定血清中12种抗癫痫药物的超高效液相色谱-串联质谱检测方法,通过梯度洗脱使12种抗癫痫药物得到较好分离;通过调整流动相,优化质谱条件,提高了检测灵敏度。采用乙腈沉淀蛋白质进行样品前处理,方法学考察回收率、精密度均满足测定需求。该方法简便、快速,能够排除内源性杂质的干扰,准确定性和定量,便于同时筛查体内多种抗癫痫药物,适用于临床上抗癫痫药物血药浓度的监测。
