基于分层矩阵能量谱的个体拷贝数变异检测算法
2020-07-23陈念华袁细国
陈念华 袁细国
(西安电子科技大学 计算机科学与技术学院,陕西 西安 710071)
0 引言
癌症对人类的健康和生命威胁极大,从基因分子水平上研究癌症的预防和治疗策略是当代医学急需解决的问题.近年来国际生物医学界广泛关注的一种新的基因组变异形式:拷贝数变异(copy number variation,CNV),为此提供了新的线索和思路.CNV 是一种基因组结构性变异,主要表现为长度从几 Kb 至 Mb 的染色体片段的扩增或缺失[1, 2],是促使人类个体间基因差异的重要因素之一,也是引发癌细胞产生和发展的重要现象.CNV扩增是指基因组区域的拷贝数从正常细胞二倍体到多倍体的变化,CNV缺失是基因组区域中拷贝数减少的变异.虽然CNV发生的频率较低,但累积的碱基数量却大大超过了单核苷酸多态.在癌细胞中,CNV变异通常会引起相应区域中包含的基因的剂量变化,这会影响基因的正常功能[3, 4].因此,在癌症基因组中CNV的准确检测对于癌细胞发展机理研究及癌症诊断具有重要的现实意义[5, 6].
以多样本数据为背景的CNV检测与分析,其过程不仅涉及到癌症样本与正常样本信号的比较,而且涉及到癌症样本本身之间的比较,那么依据CNV在样本中出现的频率,可将其分为复发性和个体性CNV模式.复发CNV指在多数样本中共同发生的CNV区域,即CNV在多样本中表现的频率较高,目前相关检测方法的研究非常丰富[7, 8].个体CNV指在少部分样本中共同发生或个体特异性的CNV,即CNV在多样本中表现的频率较低[9].而目前为止,针对个体性CNV检测的研究方法较少,但这种CNV模式同样非常重要.通过研究个体CNV与癌症的关系,不仅可以发现更多与癌症发生发展密切相关的变异,还对在医学上进行个体化的有针对性的药物开发和治疗有极大的帮助.
因此,本文提出一种名为IndivCNV(An individual copy number variation detection algorithm based on hierarchical matrix energy spectrum)的算法,与现有方法相比,该算法主要具有3个特点:(1) 可以从原始数据中实现个体性CNV模式的检测;(2) 通过全变分将观察到的信号进行平滑处理,利用潜变量模型将其重建为特征与权重的乘积,以应对噪声较高情况下CNV的检测;(3) 对信号进行分层,根据分层矩阵能量谱在每层的占比,将能量高的复发CNV信号层剔除,以更准确鉴别个体性CNV.
1 相关工作
基于阵列的比较基因组杂交技术(array-based comparative genomic hybridization, aCGH)是一种高通量、高分辨率的方法,可以用于测量数千个DNA区域中拷贝数的变化.要从aCGH数据中检测CNV,就必须定位信号数据中CNV区域与非CNV区域间的变化点,这些变化点会将染色体分成多个离散的片段,进一步便可以检测出CNV.多样本CNV的检测涉及多个样本,以期发现那些单样本检测无法发现的模式.目前有许多相关方法可以对aCGH数据进行多样本CNV检测,例如PLA(Piecewise-constant and low-rank approximation for identification of recurrent copy number variations)[10]、fastRPCA(A fused lasso latent feature model for analyzing multi-sample aCGH data)[11]、FLLat(A variational approach to stable principal component pursuit )[12]等.
PLA将多样本CNV检测问题转化为矩阵分解问题,其中原始数据矩阵被分解为低秩分量、稀疏分量和噪声分量.这三个成分分别对应于复发CNV、个体CNV和随机噪声.通过主成分分析,也就是计算出输入矩阵的奇异值分解,并使用前几个奇异向量形成一个新的低秩矩阵,可以很容易地从低秩分量中识别出复发性CNV,从稀疏分量中识别出个体CNV.
类似地,fastRPCA采用线性叠加的模型,为稳定主成分跟踪(stable principal component pursuit, SPCP)引入了新的凸公式,将原始信号分解为低秩分量和稀疏分量.fastRPCA首先建立了一个凸变分框架,然后用准牛顿法对其进行加速,并使用此创新设计了通过变分框架的快速方法.用aCGH数据作为原始输入,经过以上处理,便可以从低秩分量中识别出复发性CNV,从稀疏分量中识别出个体CNV.
FLLat使用潜在特征模型对aCGH数据进行建模,其中每个样本均通过固定数量的特征的加权组合来建模.这些特征代表了样本组CNV的关键区域,并与权重相结合,描述了每个单独样本中的CNV区域.FLLat在特征的估计中使用了融合最小绝对值收敛和选择算子,这在估计中既保证了数据的平滑度,也保证了数据的稀疏性.
以上这些方法虽然能较好的从多样本数据中检测出CNV,但是都不能对个体CNV进行针对性的检测,因此本文提出了可以对个体CNV进行针对性检测的算法IndivCNV.
2 方法
IndivCNV算法的基本框架如图1所示,其输入数据格式为大小为L×S的矩阵X,其中L代表探针数,S代表一组数据中包含的样本个数.该算法通过以下5个主要步骤实现对个体CNV的检测:(1) 基于全变分正则化的信号层次化分解,(2) 应用融合最小绝对值收敛和选择算子,(3) 计算约束权重与特征数量J,(4) 模型参数估计,(5) 用分层矩阵能量谱识别个体CNV,下面将会针对每一个步骤的相关理论和实现过程进行详细阐述.
2.1 基于全变分正则化的信号层次化分解
本文使用潜在特征模型来模拟多样本数据,并且提出逐层分解信号的策略,通过将CNV的原始数据重建为不同特征模式的组合来发现原始数据中的CNV模式.将两个秩为j的矩阵相乘的形式用j个秩为1的列向量与行向量相乘的加和来等价表示,以此来表示原始矩阵的分层分解,即
(1)

该模型说明了样本组的CNV的重要特征是由J个特征共同总结的.具体来说,每个特征代表CNV的特定模式.然后,给定样本的权重确定每个特征对该样本的贡献程度.换句话说,通过这些特征的权重可以知道不同特征的发生频率,以此来推断复发 CNV和个体 CNV.
2.2 应用融合最小绝对值收敛和选择算子
CNV区域倾向于在整个染色体的连续区域中发生,区域具有相同的拷贝数.对于未显示CNV的染色体的其余部分,预期的信号强度应为零.因此,如果我们将生物芯片数据视为沿着染色体的1维信号,则信号的大部分都为零,非零区域出现在平滑区域中.通过这种1维信号的稀缺性和平滑性的组合可以自然地想到融合最小绝对值收敛和选择算子信号近似器(fused lasso signal approximator,FLSA[13]).FLSA可以解决优化问题
(2)
其中u=(u1,…,up)T是估计所述有序结果的参数的向量.第一个惩罚项负责惩罚每个参数大小,这可以促使解决方案稀疏,第二个惩罚项负责惩罚相邻参数之间的绝对差异,这可以促使解决方案平滑.有2个相应的调谐参数,λ1和λ2,分别控制稀疏性和平滑性.

(3)
2.3 约束权重与特征数量 J
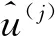
(4)
约束(4)对每行V的大小设置了限制,即对应于给定特征的权重.在此认为这是限制权重大小的最合适方式.首先,它使估计的特征之间的直接比较更有意义;其次,它可以防止大部分权重仅分布在少数几个特征上.
模型(1)中需要对特征J的数量做出选择.从理论上讲,J可以取{1,2,…,S}中的任何值,其中S是样本数.J的最好的选择对于任何给定的数据集都是难以确定的,并可能取决于许多因素,例如,噪声的水平,调谐参数λ1和λ2的值,以及S的值.因此,J的值通常留给用户指定,默认设置为min {15,S/2}.本方法也提供选择J的半自动过程,这是基于解释的变化百分比(PVE).对于给定的J值,PVE被定义为
(5)
2.4 模型参数估计
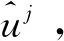
(6)
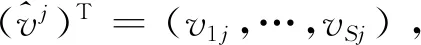
(7)
2.4.2 选择融合最小绝对值收敛和选择算子调谐参数λ1和λ2.通常,给定模型的最佳调谐参数的选择都是一个困难的任务,并且随着调谐参数数量的增加会更加复杂.为了简化对最佳调谐参数的搜索,本方法通过引入λ0和α∈(0,1)来重新定义参数λ1和λ2,使得λ1=αλ0,λ2=(1-α)λ0.在此可以认为λ0是整体调谐参数,它和α一起确定对稀疏度与平滑度的重视程度.通过固定α可能采取的值,可以有效地将对两个参数λ1和λ2的搜索简化为仅对一个参数λ0的搜索.
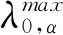
(8)
2.5 分层矩阵能量谱
(9)
(10)
其中T代表设定的占比阈值,I是大小为L×S的矩阵,代表个体 CNV.得到最终的个体CNV矩阵I以后,需要按照样本将数据区分为S个大小为L×1的矩阵,每个矩阵代表每个样本的结果.此时,需要再选定一个阈值H,若某探针处的绝对值大于H,则认为该处有个体CNV,反之则认为是正常.因为个体CNV在样本间有很大的差异,所以需要按上述对每个样本的结果数据都分别判断.
3 实验结果
3.1 模拟数据
3.1.1 模拟数据介绍. 为了评估IndivCNV算法对个体CNV的检测性能,本节将采用模拟数据进行实验,并与三种现有方法(PLA、FLLat、fastRPCA)进行比较.在文献[15]里,详细地定义了六种不同的复发 CNV场景.在本文的研究里,将采用这六种场景来生成模拟数据.在每一种场景下生成50组数据,每组数据是50×5000的矩阵,其中50代表50个样本,5000代表每个样本上的5000个探针.在生成每组数据时,无CNV区域的信号值设为0;复发 CNV区域位于探针1876到3125之间,其模式参考图2,将缺失变异区域的信号值设为-1,扩增变异区域设为1.每个样本还需要在不与复发 CNV区域重合的部分,随机选取一个位置,添加一个长度为500探针的个体 CNV,个体 CNV的信号值从{-2,-1,1,2}中随机选取,最后再向整个数据加入高斯噪声.
6种不同场景生成模拟数据的过程展示在图3,图中黄色区域代表扩增,蓝色区域代表缺失.其中第一行是根据文献[15]中对不同场景的描述生成的只有复发 CNV的数据,第二行是在复发 CNV的基础上随机添加个体 CNV的数据,第三行是添加了噪声水平为1的高斯噪声的最终模拟数据.每组数据的纵向代表样本,横向代表探针.从图上可以看出,这六种场景可以分为两类,场景1、3、5为一类,它们只有一个复发CNV区域;场景2、4、6为一类,它们含有多个复发CNV区域.本文的研究任务是从这些最终的模拟数据里准确恢复出个体 CNV.
3.1.2 检测结果热图展示. 在图4中展示了在6种场景下不同方法对个体 CNV的检测结果.从图中可以看出来IndivCNV检测出了绝大部分的个体 CNV,并且能很好地把个体 CNV与复发 CNV区分开来,没有将复发 CNV误判为个体 CNV.FastRPCA可以分辨出一部分个体 CNV,但是没有将噪声很好地剔除,因此难以识别检测出的个体 CNV的模式;而PLA则倾向于将一个完整的个体 CNV切割成多个小段,有明显的缺失; FLLat的特点是它做检测时不对复发 CNV与个体 CNV进行区分,导致两种类型的CNV都存在于结果数据中.由以上分析可知IndivCNV在检测个体CNV时确实更加有优势,但是从图中可以看出它还是存在一定的缺陷,因为它更趋向于检测出发生个体CNV频率较高的位置的变异,而对于发生频率小的个体CNV,则很难检测出.
3.1.3 检测结果ROC曲线. 为了可量化地评估这些方法,本研究进一步通过ROC曲线评估各方法在六种场景下的个体 CNV识别性能.ROC(receiver operating characteristic curve)是一种显示分类模型在所有分类阈值下的效果的图表,其横轴是假阳性率 (False Negative Rate,FPR),纵轴是真阳性率 (True Negative Rate,TPR).FPR指的是所有非个体 CNV区域中被误判为个体 CNV的比率,该值越小越好,TPR指的是在所有检测出来为个体 CNV的区域里,确实是个体 CNV的比率,该值越大越好.ROC曲线的作用在于,在很多分类器分析中,得到的预测值通常不是0或1,而是一个0-1之间的概率值,此时就需要人为设定一个阈值,比如设定大于0.6则为1,反之则为零.但是不同的阈值所带来的预测结果一定有差异,此时就可以用ROC曲线来刻画不同阈值给分类器带来的影响.通过上文对FPR和TPR含义的介绍可知,ROC曲线越靠近左边沿和上边沿,说明模型越好,因为此时TPR足够大,FPR足够小,说明分类的正确率很高.而ROC曲线上不同的点对应着模型对不同阈值的预测水平,简单来讲,阈值越大,点越靠近左下,反之越靠近右上.
图5展示了各方法在6种场景下的ROC曲线.这些ROC曲线是通过对各方法检测出来的结果数据设定不同的阈值生成的.从图上可以看出, IndivCNV检测个体 CNV的性能优于其他三种方法.例如在场景1的ROC曲线中,当FPR=0.1时,IndivCNV的TPR就已达到0.8,而FLLat的TPR只有0.45,PLA和fastRPCA的TPR仅有0.3;在场景2中,虽然当FPR值大于0.3时,FLLat和IndivCNV的曲线基本重合,但是IndivCNV在FPR=0.05时TPR就已经达到了0.7,这说明IndivCNV在低FPR水平就可以实现较高水平的TPR;在场景3、5、6中,呈现出同样的趋势:当FPR较高时,FLLat与IndivCNV的曲线十分接近,但是始终都低于IndivCNV,只有在场景4中曲线的后半段FLLat超过了IndivCNV,尽管如此,其前半段依旧远低于IndivCNV的ROC.
综上所述,与fastRPCA和PLA相比,IndivCNV和FLLat算法对个体CNV的识别结果具有更高的TPR.然而,FLLat的性能与IndivCNV虽然较为接近,但仅表现在FPR较高的情况,当FPR较低时,其ROC曲线依旧远低于IndivCNV.因此,在对个体CNV的检测中, IndivCNV算法具有更明显的优势.
3.2 真实数据
为了证明IndivCNV在真实数据上的可用性,本实验引入异质性乳腺癌CNA真实数据集对算法进行验证.这个数据集中包含了112个乳腺癌样本的SNP array数据,每个样本都有23条染色体上的不同数据,每条染色体的探针各不相同,由Illumina 109 K SNP array平台采集.在进行实验时,首先将每个样本不同染色体上的数据分割开来,然后将处理所得的CNV分段在基因组区域对齐,成为一个大小为112×pi的变异强度矩阵,其中112代表样本数,pi代表在第i条染色体上的探针数,即分割完成后有22个变异强度矩阵(因为乳腺癌是常染色体上的疾病,所以仅对前22条常染色体进行实验),并分别对这22个信号矩阵进行实验分析.在实验过程中,使用IndivCNV对数据进行分析,阈值T设为0.1.为了消除每个样本中的波谱偏差,需通过局部中值减去信号数据,中值计算的窗口大小是染色体长度的四分之一.
对于IndivCNV算法在乳腺癌数据中所发现的个体CNV区域,本研究通过乳腺癌相关文献报道的CNV区域对算法结果进行验证.对于IndivCNV算法所发现的个体CNV区域,其中许多区域被现有文献报道为乳腺癌CAN驱动区域.例如,IndivCNV算法成功识别出17号染色体上的ERBB2基因[16],该基因曾被多项研究报道为乳腺癌CAN驱动变异.同时,IndivCNV在14号染色体发现AKT1基因[17],而该基因则被报道与乳腺癌的发生发展密切相关.表1汇总了IndivCNV所发现的个体CNV与现有文献报道发现与乳腺癌有密切关系的基因重合的结果.上述结果表明,IndivCNV算法所发现的个体CNV区域与已报道CNV驱动变异区域具有较高的一致性.
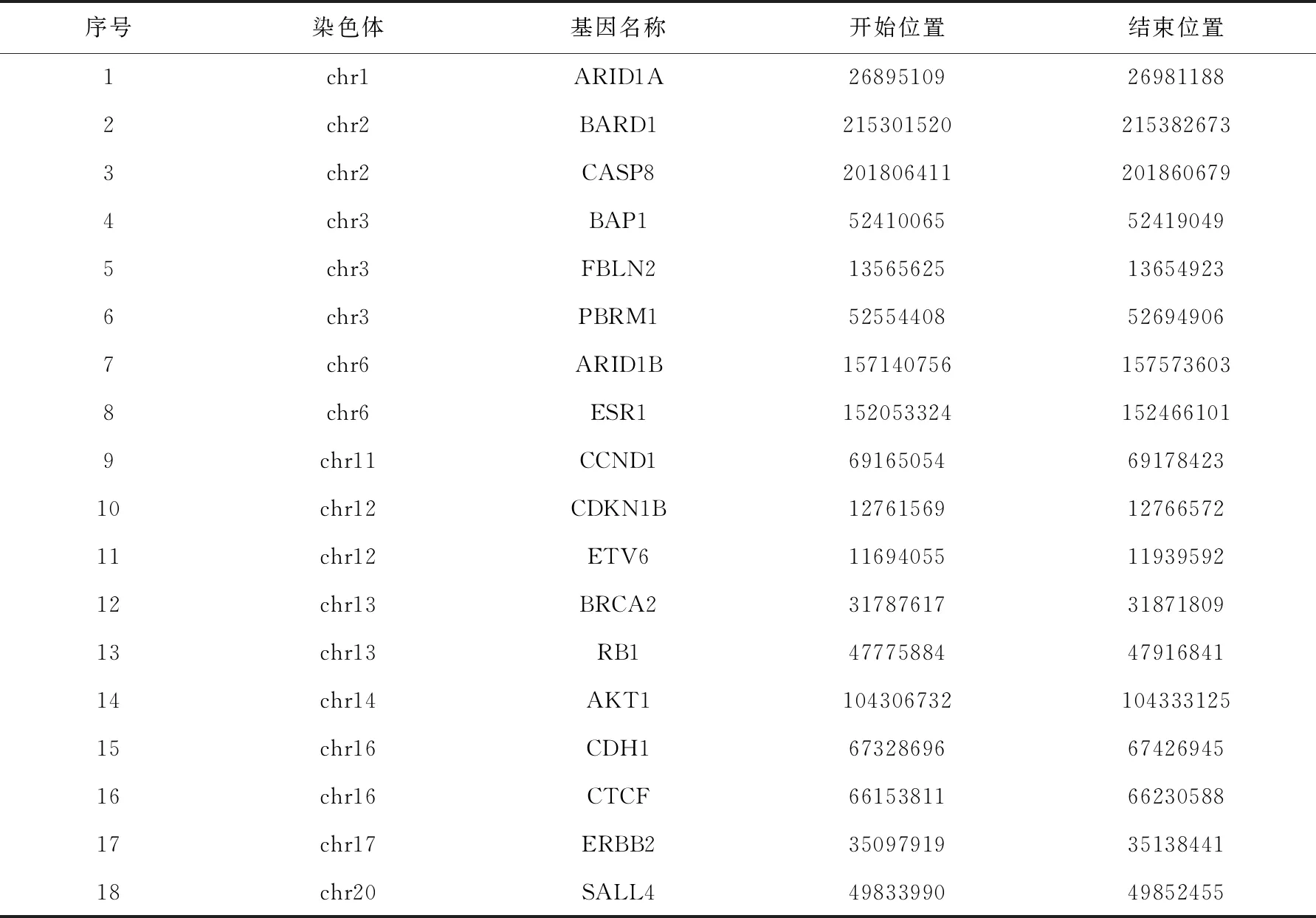
表1 IndivCNV检测出与现有文献报道发现与乳腺癌有密切关系的基因重合的结果
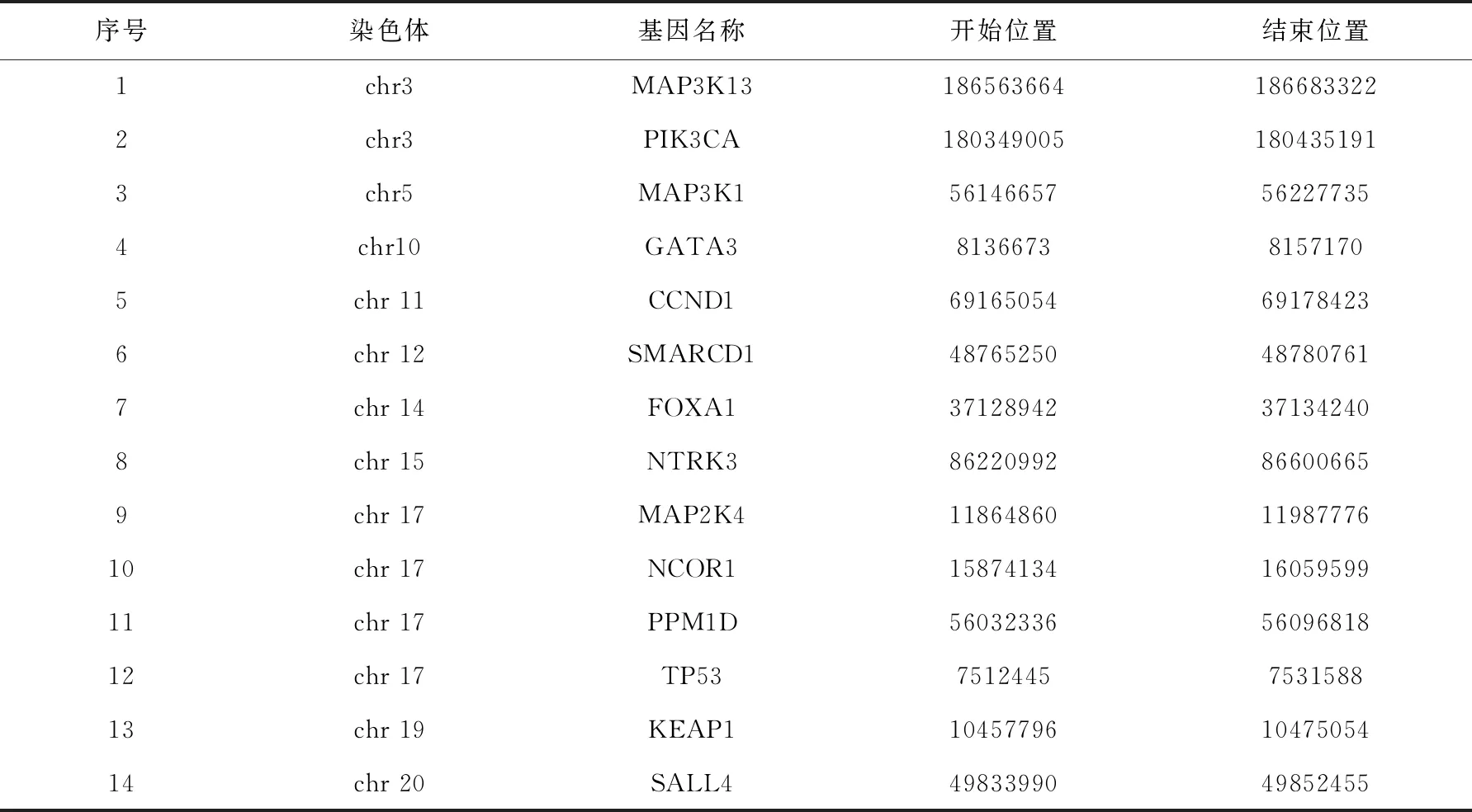
表2 IndivCNV的复发CNV模式所匹配到的乳腺癌相关基因
正如第3节所说,IndivCNV在做个体CNV模式检测的过程中,会将复发CNV的模式剔除.在此,本实验在用该真实数据检测时,将剔除的复发CNV数据也另行保存,并对复发CNV模式进行驱动基因匹配.表2中汇总了在IndivCNV的复发CNV模式中发现的乳腺癌驱动基因,表3汇总了IndivCNV在真实数据检测出的个体CNV模式在复发CNV模式之外发现的驱动基因.由表2、3可以看出,个体CNV的检测可以很大程度上弥补复发CNV对驱动基因发现的不足,例如,在表2复发CNV的检测结果里,未发现1号染色体和13号染色体上有与乳腺癌相关的基因,而在个体CNV模式里则发现了1号染色体上的ARID1A基因,13号染色体上的BRCA2基因和RB1基因,这几个基因都是乳腺癌相关基因,并被权威癌症基因数据库Cancer Gene Census所收录[18-20].上述结果表明,IndivCNV算法的个体CNV发现结果可有效弥补现有方法发现结果的不足,同时也证明了个体CNV检测对于癌症研究的重要性.
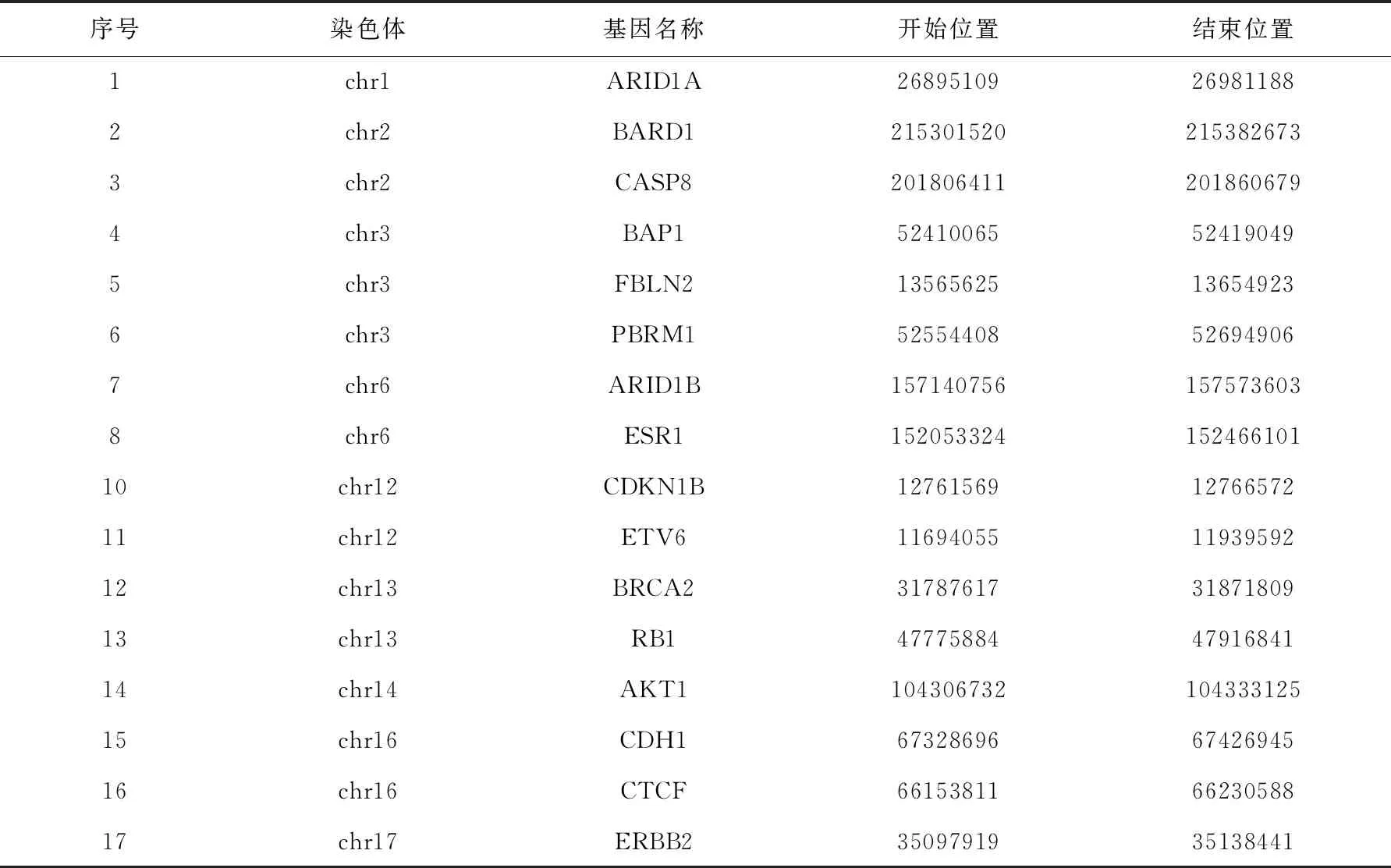
表3 IndivCNV的个体CNV模式在其复发模式之外检测到的乳腺癌相关基因
4 结论
CNV是导致癌症发生发展的重要因素之一,由于现有研究更侧重于对复发CNV的研究,对个体CNV的关注程度不够,忽略了个体CNV的研究价值,因此本文通过分析个体CNV的模式,提出了一种新的适用于发现个体CNV的算法IndivCNV.IndivCNV首先需要使原始信号趋于平滑,因此采用了全变分正则化的方式达到此目的;接着将原始数据的每个样本建模为固定数量的特征的加权和,这一步使用了潜变量模型和融合最小绝对值收敛和选择算子惩罚;然后使用信号层次化分解,将不同模式的CNV用不同层的矩阵表示;最后利用分层矩阵能量谱,根据复发CNV模式能量占比大,个体CNV模式的能量占比小的原理,将复发CNV与个体CNV区分开来,最终达到检测个体CNV的目的.
在本文的实验中,首先将IndivCNV应用到六种不同场景的模拟数据上,同时将fastRPCA、PLA、FLLat这三种算法也应用到该模拟数据上,以ROC曲线为性能判断标准,根据检测结果选定不同阈值绘制ROC,以此进行性能对比,实验结果表明,IndivCNV检测个体CNV的性能显著高于已有的三种方法的性能.然后又使用IndivCNV检测异质性乳腺癌CNA真实数据集中的个体CNV,检测个体CNV结果中包含许多现有文献已报道过与乳腺癌相关的基因,还发现了复发CNV模式没有发现的与乳腺癌相关的基因,因此IndivCNV的性能在实际数据上也得到了验证.综上所述,IndivCNV在个体CNV方面的检测性能确实有了大幅提升.
