团簇NiMo3P 的磁学性质研究
2020-07-18王美玲方志刚赵振宁
王美玲,方志刚,赵振宁,秦 渝
(辽宁科技大学化学工程学院辽宁鞍山114051)
团簇磁性是目前团簇的研究热点之一,人们希望通过对团簇磁性的研究合成新的磁性材料,这对工业和军事等方面都具有重要意义[1].过渡金属团簇不仅具有良好的磁学性质,还具有非常优良的催化性、耐腐蚀性、耐磨性等等[2-3].近年来环境问题日趋严峻,人们的环保意识显著增强,国家对汽车尾气的排放也做出了限制[4].煤焦油加氢脱硫是制取燃料油的主要方法之一[5],为了减少有害气体的排放,需要研制出更加高效的加氢脱硫催化剂. Varsha Jain 等人[6]基于DFT 计算,研究了双金属磷化物作为催化剂时苯酚的加氢脱氧反应,结果表明NiMoP 催化剂作用于苯酚的加氢脱氧反应分别通过环加氢脱氧和环加氢脱水途径生成环己烷和环己烯,为NiMoP 催化剂催化煤焦油化合物的具体反应途径提供了依据.NiMo 系列催化剂是目前工业上应用最广泛的加氢脱硫催化剂之一[7],本文引入非金属原子P 进行掺杂,以团簇NiMo3P 为模型进行研究[8].磁性催化剂具有容易分离回收利用等特点[9],运用磁性载体进行催化可以大大节约运行成本.Yong Liu 等人[10]发现Mo基化合物存在半金属态,采用FP-LAPW GGA 方法得到了 闪锌矿(ZB)结构的MoGe 和MoSn 合金,其中ZB-MoSn 为自旋无间隙半导体,ZB-MoGe 为无间隙磁性半导体,此研究为寻找居里温度较高的4d基合金提供了可能性.Dican Lu 等[11]以Ni 纳米球为初始镍源,NaH2PO2·H2O 为磷源,采用简单的低温磷化工艺,成功地制备了磁性Ni@NixPy 核壳结构,其研究结果表明Ni 纳米球表面NixPy 层的形成提高了最终产物的磁性,且Ni@NixPy 核壳结构对4-NP 的还原具有较好的催化性能,在实际应用中具有重要意义. 故本文对团簇NiMo3P 进行磁性相关的实验分析,希望可以为研制Ni-Mo-P 催化剂提供依据.
1 模型和计算方法
根据拓扑学原理,运用密度泛函理论(Density functional theory,DFT)[12],利用Gaussian09 程序对二、四重态下团簇NiMo3P 的22 种构型进行优化计算,获得稳定构型.利用Multiwfn 程序得到团簇NiMo3P各构型的原子磁矩、各原子的Mulliken 自旋布居数与s、p、d 轨道成单电子数.在B3LYP 泛函的条件下,采用Lan12dz 基组对Ni 的最外层3s23p63d84s2价电子、Mo 的最外层4s24p64d55s1价电子及P 的最外层3s23p3价电子进行描述. P 原子的核外电子排布为1s22s22p63s23p3,其价电子没有d 轨道的存在,但大量实验表明,在计算时第三周期元素存在d 轨道,其d 轨道为价轨道,参与s、p、d 杂化成键[13].本文在B3LYP/Lan12dz 水平下,对Ni、Mo 原子采用Hay 等人的18-eECP 双ξ 基组(3s,3p,3d/2s,2p,2d)[14];P原子采用Dunning/Huzinaga 双ξ 基组(9s,5p/3s,2p),且P 加极化函数ξP.d=0.55[15].以上所有计算均在启天计算机M4390 上完成.
2 结果与讨论
2.1 优化构型及稳定性
对团簇NiMo3P 的所有构型进行优化计算,在排除相同构型与不稳定构型后,得到了5 种稳定构型,其中含有4 个四重态和1 个二重态构型.根据图1所示,团簇NiMo3P 的所有优化构型均为三角双锥型,表明三角双锥型为团簇NiMo3P 的优势构型,使其具有更好的稳定性.将构型按能量由低到高依次排序,以能量最低的构型1(4)为基准,其能量为0 kJ/mol,然后依次计算出其余构型的相对能量,右上角括号内的数字表示重态. 根据能量可知,团簇NiMo3P 各构型稳定性为1(4)>2(4)>1(2)>3(4)>4(4).

图1 团簇NiMo3P 的优化构型图Fig.1 The optimized configurations of cluster NiMo3P
2.2 磁学特性
原子核外成单电子的存在使团簇产生磁性.团簇的磁性仅与成单电子数有关,因此,研究团簇的成单电子对团簇的磁性具有非常重要的意义.根据电子的自旋方向,可以将电子分为自旋向上和自旋向下两种,自旋向上为正的α 电子,自旋向下为负的β 电子.表1 中列出了团簇NiMo3P 各构型s、p、d 轨道的成单电子数,其中各构型的d 轨道数值均为正值,这说明团簇NiMo3P 各构型的d 轨道均是自旋向上的α电子,而s、p 轨道皆具有自旋向上和自旋向下两种电子. 根据表1 可以看出,团簇NiMo3P 各构型的d轨道成单电子数远大于s、p 轨道,这说明团簇NiMo3P的磁性绝大部分是由d 轨道贡献. 观察表1 可发现,团簇NiMo3P 各个四重态构型的成单电子数远大于二重态构型1(2),即二重态构型1(2)的磁性较四重态来说更小.二重态构型的成单电子数为1,四重态构型的成单电子数为3,这与自旋多重度的定义相一致[16].
图2 为团簇NiMo3P 各构型原子磁距的变化趋势图,由图可知,除二重态构型1(2)外,其余构型的Mo原子磁矩均大于零,说明Mo 原子的引入使团簇NiMo3P 的磁性增加;而构型1(2)的Mo 原子磁矩小于零,这也侧面印证了二重态构型的磁性较四重态来说更小这一论点.在构型1(2)中Ni 原子的存在对团簇NiMo3P 的磁性起主要贡献作用,除构型1(2)之外,在其余构型中Mo 原子的磁距值较大(分别为3.99 μB、2.26 μB、1.87 μB、和1.88 μB),表明在这四个构型中Mo 原子对团簇NiMo3P 的磁性贡献最大. 对于Ni原子来说,除构型1(4)的磁距为负值外,其余构型均为正值,所以Ni 原子的引入也使得团簇NiMo3P的磁性增加.且Ni 原子磁距的变化趋势与Mo 原子恰好相反,构型1(4)—1(2),Mo 原子的磁距随构型能量的增加而减小;从构型1(2)—4(4),随着构型能量的增加Mo 原子的磁距也相应的增加;但Ni 原子的磁距随着构型能量的变化却呈现先增加再减少的趋势,表明Ni 原子与Mo 原子之间可能存在拮抗作用,呈现出此消彼长的效果. 从图2 中可以看出,Mo原子与Ni 原子的变化幅度都较为剧烈,而P 原子的变化幅度较小. 构型1(4)和2(4)P 原子的磁距为负值,其余三个构型的磁距也较小(分别为0.016 μB、0.188 μB 和0.186 μB),这说明P 原子的引入会降低团簇NiMo3P 的磁性. 这验证了徐诗浩在《团簇V3B2成键及磁学性质研究》中提出的金属原子的引入会增加团簇磁性,而非金属原子的掺杂会降低团簇磁性这一观点[17].
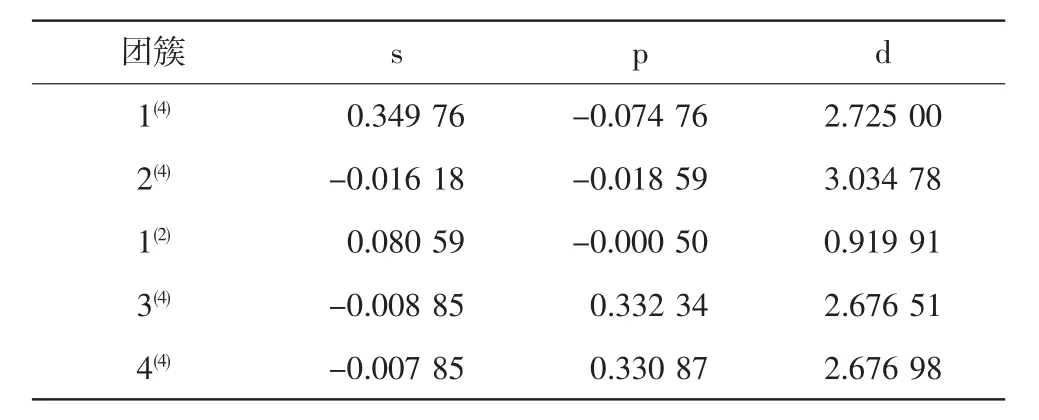
表1 团簇NiMo3P 各构型的s、p、d 轨道成单电子数Tab.1 The s,p,d orbitals of the various configurations of the cluster NiMo3P form a single electron number

图2 团簇NiMo3P 各构型的原子磁矩Fig 2 Atomic magnetic moments of various configurations of clusters NiMo3P
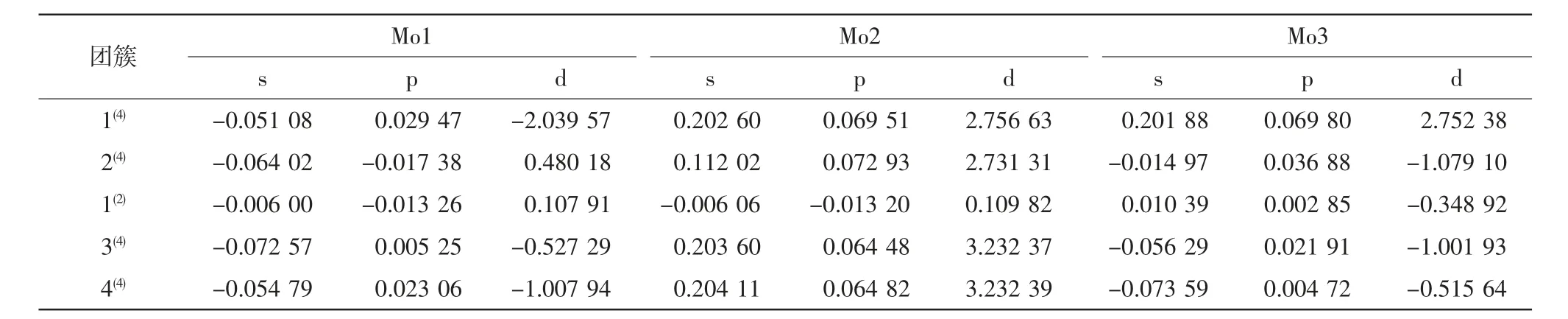
表2 团簇NiMo3P 各构型Mo 原子的Mulliken 自旋布居数Tab.2 Mulliken spin population of Mo atoms with different configurations of cluster NiMo3P

表3 团簇NiMo3P 各构型Ni 原子和P 原子的Mulliken 自旋布居数Tab.3 Mulliken spin population of Ni and P atoms in clusters NiMo3P
表2 和表3 为团簇NiMo3P 各构型原子的Mulliken自旋布居数,自旋布局表示某片段或某原子轨道所具有的成单电子数,原子轨道的自旋布居数加和即为原子的自旋布居数,各原子的自旋布居数加和即为分子的自旋布居数.从上表可知Mo 原子的s、p、d三个轨道中,d 轨道具有最多的成单电子,即d 轨道为Mo 原子磁性的主要贡献者.在团簇NiMo3P 的三个Mo 原子中,除磁性最小的构型1(2)外,Mo2 的s、p、d 三个轨道均为正的α 电子,且提供的成单电子数最多(1(4):d:2.75663;2(4):d:2.73131;3(4):d:3.23237;4(4):d:3.23239),故Mo2 为团簇NiMo3P 磁性的主要贡献者.Ni 原子与Mo 原子情况相同,亦是d 轨道为Ni 原子的磁性提供了最多的成单电子.P 原子提供的成单电子较少,且集中在p 轨道上.构型1(2)的Ni原子s、p、d 轨道均为自旋向上的α 电子,且成单电子数远大于Mo 原子与P 原子(Mo1:d:0.10791;Mo2:d:0.10982;Mo3:d:-0.34892;Ni:d:1.05134;P:p:0.01566),故构型1(2)的磁性主要由Ni 原子的d 轨道提供.其余构型Mo 原子的d 轨道均具有最多的成单电子,故除1(2)外的其余构型,磁性均主要来自Mo原子的d 轨道.
图3 给出了团簇NiMo3P 各构型s、p、d 轨道的态密度图像,不仅可以更加直观的观察各构型s、p、d轨道对团簇磁性的贡献情况,还可以在图中观察到团簇NiMo3P 各构型s、p、d 轨道上α 电子与β 电子的电子数目与每个能级上α、β 电子的具体分布情况.图中实线表示自旋向上的α 电子,虚线表示为自旋向下的β 电子,其中虚线所表示的自旋向下的态密度曲线积分为负值,故虚实两条态密度曲线的积分之和即为所剩的成单电子数.团簇态密度图像对称性越好,则剩余的成单电子数越少,团簇磁性越小.从图3 中可以看出,构型2(4)、3(4)、4(4)的s、p 轨道对称性较好,这就表明在s、p 轨道上剩余的成单电子数较少;观察这三个构型的d 轨道,可以看出在C 和C’处的波峰差异较大,剩余的成单电子较多,即对于这三个构型来说d 轨道对团簇的磁性起主要贡献作用,s、p 轨道贡献较小,与表1 得出的结论一致.观察构型1(4)的态密度图像,可以看出p 轨道的两条曲线相互对称;而s 轨道和d 轨道中A、B、C 和A’、B’、C’处的波峰差异较大,即1(4)构型的s 轨道和d 轨道都对团簇的磁性起到一定的贡献作用,s 轨道对团簇磁性的影响不可忽略.对于构型1(2)来说,其s、p、d 轨道的态密度图均具有良好的对称性,故构型1(2)的成单电子较少,导致该构型磁性较小,验证了表1 的分析结果.观察图3,可以看到构型3(4)与4(4)s、p、d 轨道的态密度图近似相同,其各个峰的形状十分接近,仅在峰值上存在微小的差异(3(4)(s:-0.00885;p:0.33234;d:2.67651)、4(4)(s:-0.00785;p:0.33087;d:2.67698)),且构型3(4)与4(4)各个原子的磁距也近似相等(3(4)(Mo:1.86952;Ni:0.94278;P:0.18771)、4(4)(Mo:1.87715;Ni:0.93732;P:0.18554),说明构型3(4)、4(4)的磁学性质相近.由于电子自旋的不确定性,会导致α、β电子部分发生翻转.从图中可以看出,各构型的s 轨道上10~20 eV 范围内α、β电子发生自旋方向的改变;在0 eV 处,仅磁性最小的构型1(2)的电子发生翻转. 团簇NiMo3P 的p 轨道上,10 eV 处各构型均发生了电子的翻转. 而在d 轨道上,仅有构型1(2)在0~10 eV范围内电子自旋方向改变.

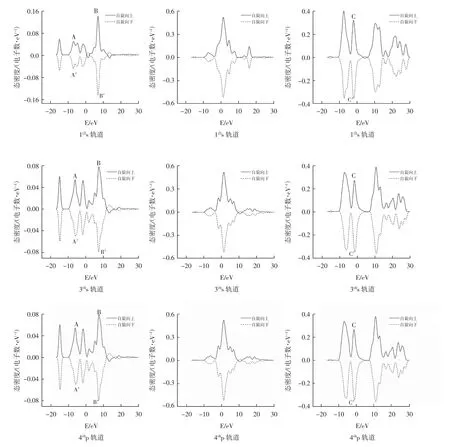
图3 团簇NiMo3P 各构型s、p、d 轨道的态密度分布Fig.3 Density distribution of states of s,p and d orbitals of various configurations of NiMo3P
3 结 论
本文结合团簇NiMo3P s、p、d 轨道的成单电子数、态密度分布和各原子的磁距对团簇NiMo3P 的磁学性质进行了分析.分析结果表明团簇NiMo3P 具有4 个四重态和1 个二重态,5 种稳定构型,各构型稳定性为1(4)>2(4)>1(2)>3(4)>4(4),三角双锥型为团簇的优势构型.团簇NiMo3P 各构型的d 轨道成单电子数远大于s、p 轨道,即团簇NiMo3P 的磁性绝大部分是由d 轨道贡献.由于四重态构型的成单电子数较多,故四重态构型的磁性比二重态更强.Mo 原子的s、p、d 三个轨道中,d 轨道具有最多的成单电子,即d 轨道为Mo 原子磁性的主要贡献者;且三个Mo 原子中,Mo2为团簇NiMo3P 提供了最多的成单电子.除磁性最小的二重态构型1(2)由Ni 原子的d 轨道提供了最多的成单电子外,其余构型的磁性均主要来自Mo 原子的d 轨道.Mo 原子和Ni 原子的引入使团簇NiMo3P 的磁性增加,Ni 原子与Mo 原子之间可能存在拮抗作用,而P 原子的掺杂会导致团簇的磁性降低.且由于电子自旋的不确定性,会导致α、β 电子的自旋方向发生改变.
