辣椒雄性不育分子生物学研究进展
2020-07-14SaleshKumarJindalMajorSinghDhaliwalOmPrakashMeena
Salesh Kumar Jindal Major Singh Dhaliwal Om Prakash Meena
(1. Department of Vegetable Science, Punjab Agricultural University, Ludhiana 141004, India;2. Directorate of Research, Punjab Agricultural University, Ludhiana 141004, India)
1 引言
辣椒原产南美,有34个栽培种和野生种[1-4],多数为二倍体,2x = 24。辣椒独具辣味,辣味强度由辣椒碱含量决定,辣椒素是一种主要的辣椒碱类物质[5]。目前世界上栽培的辣椒品种,主要属于C. annuumL.、C. chinenseJacq.、C. baccatumL.、C. frutescensL.和C. pubescensRuiz & Pavon等 5个栽培种。C. annuum种植广泛,并驯化和选育了大量的品种,一般有辣味辣椒作为香料,甜椒作为蔬菜[6]。辣椒适应性强,在热带和温带地区广泛种植,甜椒则在温带地区更受欢迎。辣椒为常异花授粉作物,杂种优势非常明显。近年来,优良杂交品种因受农民青睐而大面积推广。利用雄性不育系生产杂交种子,可降低制种成本50%左右,提高种子纯度[7]。辣椒NMS和CMS均在杂交种子生产上应用。
Martin 和Crawford(1951)首次在灌木辣椒种发现核雄性不育[8]。迄今为止,已报道20个独立遗传的核不育基因,即ms1(与msp等位基因)、ms2到ms9(mc9)、ms10(mc509)、ms11(mc705)、ms12到ms15、msc-1、msc-2和ms k[6,9-11]。除基因Dms(ms5突变)为显性外[12],其他核不育均由一个单隐性基因控制。核不育基因之间关系尚不完全清楚,Lee等[13]报道认为ms1、ms3、ms10和msk基因彼此为非等位基因。
Peterson(1958)在印度引入的“PI 164835”材料中首次发现胞质雄性不育(CMS)[14]。研究表明,CMS不育由线粒体基因组决定,母系遗传,不育性是由细胞质不育(S)和细胞核不育(rf)相互作用决定,不育性通过基因(Rf)恢复;除了遗传因素外,低温也会引起育性暂时恢复[11,15]。不育系(A系)基因型为Srfrf,保持系(B系)基因型为Nrfrf,恢复系(C系)基因型为NRfRf,可利用CMS“三系”生产杂交种子。
利用NMS和CMS生产杂交种子,各有利弊。利用不育系选育辣椒新品种,关键是选育不育系、保持系和恢复系。利用NMS缺点:传统的多代回交方法选育NMS系,繁琐、费时[9];生产种子时母本中只有50%不育株。优点:不要保持系,通过不育系后代中可育株与不育株杂交生产不育系;环境因素对不育性影响较小;容易找到恢复系。因此NMS在甜椒和辣椒杂交育种中应用广泛。利用CMS缺点:不育性受低温环境因素[14-15]、多等位遗传[16-17]和修饰基因[15,18]的影响,生产杂交种时容易出现微粉,影响种子纯度;恢复系较少。优点:母本100%不育,杂交种子生产过程中无需去雄,节约生产成本。
过去二十年,植物分子遗传学和DNA技术取得了重大进展。DNA标记已成为作物改良计划的重要组成部分[19-20]。基于PCR标记,辣椒已经构建了一批具有覆盖整个辣椒基因组、分布广泛的种间和种内遗传图谱,其中Qin等[4]和Kim等[21]分别于2014年发布了最全面的遗传图谱。育种家从辣椒基因组获取信息,从不同的连锁群(LGs)中选择标记,为标记辅助育种(MAB)和基因克隆提供技术支撑,有利于选育优良育种材料。
利用现有的遗传图谱,开发与NMS和CMS基因的相关标记。开发的标记,特别是共显性标记,能够区分杂合子和纯合子。利用这些标记,通过对基因型进行选择,能缩短育种时间。连锁标记有助于快速筛选和挖掘等位基因。种质资源筛选表明,辣椒中Rf基因较多,甜椒中rf基因较多[22-23]。通过MAB方法,将辣椒的“Rf”等位基因转入甜椒C系,将甜椒的“rf”等位基因转入辣椒B系,可扩大CMS系应用范围;利用恢复性状的修饰基因相关标记,可选育高恢复率的C-系[18]。利用NMS连锁标记,提高不育系的利用效率,确保亲本纯度100%,降低种子生产成本,提高种子产量[24]。近年来,品种的DNA指纹图谱已成为品种登记的强制性要求,也是保护育种者权利的必要手段[25]。
本文收集了与辣椒雄性不育相关的最新标记信息,并推测它们在辣椒杂种优势利用与遗传育种中的应用前景。
2 构建遗传图谱
分子标记技术促进了作物遗传连锁图谱的构建、遗传分析和分子辅助选择。等位基因挖掘、基因定位和克隆,需要标记重复性好的遗传图谱[26]。
Tanksley(1984)[27]在14个同工酶的基础上,构建了第一张辣椒分子连锁图谱,其中9个同工酶位点排列在4个LGs中。Prince等[28](1993)基于192个RFLP标记,构建了一张由19个LGs组成、遗传距离为720 cM的连锁图谱。Livingstone等[29]基于特定的F2群体构建的遗传图谱,由11个大LGs(76.2~192.3 cM)和2个小LGs(19.1~12.5 cM)组成,图谱距离1 245.7 cM,标记间的平均距离9.0 cM。由于许多标记以番茄基因为探针,因此利用该图谱可以对辣椒基因组与番茄基因组进行比较。
Kang等[30]用 TF68(C.annuumcv.)×Habanero(C. chinensecv.)的F2群体,构建了辣椒种间连锁图,含有150个RLFP和430个AFLP标记,由11个主要LGs(206~60.3 cM)和5个次要LGs(32.6~10.3 cM)组成,遗传距离1 320 cM,标记间平均距离7.5 cM;80%的RFLP标记来源辣椒基因克隆,并在整个基因组中均匀分布。该连锁图谱覆盖率与Livingstone等[29]构建的相似,为研究辣椒次生代谢产物提供依据。Rao等[31]用 “Maor”(C. annuumcv.)דBG 2816”(C.frutescens野生种)的BC2群体,构建辣椒种间连锁图谱,92个RFLP标记分布在12条全长1 100 cM的染色体上,与Livingstone等[29]构建的特异性图谱相似,并对辣椒产量相关性状进行了QTLs定位。
Lee等[32]构建了“SNU2”辣椒遗传图谱,由15个连锁群组成,共有333个标记,包括46个SSR和287个RFLP标记(146个新位点和141个来自Kang等[30]开发的SNU-图谱),覆盖距离1 761.5 cM,标记间平均距离为5.3 cM。新增的146个RFLP标记位点涵盖了辣椒基因组克隆位点、辣椒抗性相关克隆位点、番茄基因组克隆位点、辣椒素相关克隆位点以及辣椒其他的一些基因位点。SNU2图谱有15.3%标记(51个标记)来源番茄,其余标记来自辣椒。
Sugita等[33]构 建 的 甜 椒 DH系(K9-11×AC2258)连锁图谱,含有518个标记(382个AFLP、122个RAPD、7个SCAR、3个 RFLP和4个CAPS标记),由11个大LGs(56.7~118.5 cM)和5个小LGs(1.8~33.1 cM)组成,图谱距离1 043.1 cM,标记间的平均距离为4.6 cM。Ben-Chaim等[34]用“Maor”和“olyminary”的杂交后代构建了辣椒-番茄比较遗传图谱, RAPD、RFLP和AFLP等177个标记分布在12个连锁群中,全长1 740 cM。RFLP标记的排列顺序与Livingstone等[29]报道的相似,并首次鉴定了与辣椒果实性状相关的55个QTLs。Ben-Chaim等[35]基于SSR、AFLP和RFLP标记(728个标记)构建了另一张连锁图谱,由12个大LGs和4个小LGs组成,总长度1 358.7 cM,鉴定了与辣椒素含量相关的QTLs。
Barchi等[36]用“Yolo Wonder”和“Criollo de Morelos 334”(CM334)亲本的重组自交系群体,构建了一张高分辨率连锁图谱,包含507个AFLP、40个SSR、19个RFLP、17个SSAP和4个STS标记,图谱总距离1 857 cM,其中1 553 cM定位到12个辣椒单倍体染色体上,还有304 cM未定位。在49个LGs中,14个LGs包含10~60个标记,36个LGs包含<10个标记,标记间平均距离为5.71 cM。该图谱比Ogundiwin等[37]报道的图谱长(1 466 cM)。
Yi等[38]构建的连锁图谱,含有139个新的EST-SSR、41个已发表的SSRs(共180个SSRs)和63个RFLP标记组成,有14个LGs,距离为2 201.5 cM,标记间的平均距离为9.1 cM;EST-SSR标记分布在所有LGs上,每条LG有2~39个ESTSSRs。由于相互易位造成的假连锁,图谱中1号和8号染色体没有分开,因此没有8号染色体[29,32]。Lee等[32]将13号LG和15号LG分别定位到2号和12号染色体上,3个小LGs没有定位。
Minamiyama等[39]利用DH群体构建的C.annuum连锁图,包含374个SSR、AFLP、CAPS和RAPD标记,其中106个新开发的SSR标记分布在13个LGs中,覆盖1 042 cM的距离。除13号LG外,所有LG均含有2个以上的SSR标记,其中C位点被定位到13号LG上。PIC值较高的标记均匀定位到LGs上,该连锁图谱可作为辣椒的参考图谱。
目前辣椒还没有一张完整的基因组遗传图谱,12个连锁群不能对应于单倍体染色体。由于不同图谱中共同标记较少,图谱之间的标记不能比较。因此,需要一张形成共识的基因图谱作为参考遗传图谱,不同连锁群有稳定的标记,能提供综合信息。
Lefebvre等[40]利用3个DH群体构建的种内遗传图谱,有14个LGs,含有85个RFLP和RAPD标记,覆盖距离820 cM。Lefebvre等[26]报道的第一个功能详细图谱,由3个单独的种内图谱排列而成,长685~1 668 cM,有16~20个LGs,其中包括已知功能基因。这3个图谱的整合使得12个LGs与辣椒的染色体相对应,显示出与番茄图谱的对应关系。共有100个已知功能基因标记和5个具有经济价值的位点。
Paran等[41]利用6张独立图谱构建的辣椒综合图谱,由2 262个标记组成,覆盖长度1 832 cM,主要包括AFLP(1528)、番茄和辣椒RFLP(440)、RAPD(288)和一些已知的基因序列、同工酶和形态学标记。综合图谱合成了15个gaps,而单个图谱的gaps数量从21到50不等。综合图谱的平均标记密度为0.8 cM,最大密度为2.1 cM。综合图谱有助于增加标记密度和基因组覆盖率,开发紧密连锁标记,支撑MAS在育种中应用。Lee等[42]用4张图谱[32,38,43-44]构建的另一张辣椒综合图谱,含 有 AFLP、RFLP、rRAMP、SSR、BAC-end序列STS、RFLP序列STS和WRKY等标记,有805个种间标记(密度2.2 cM,图谱距离1 858 cM)和745个种内标记(密度2.5 cM,图谱距离1 892 cM),新开发的单拷贝型PCR分子标记132个(57个来自BAC末端序列,13个来自RFLP和62个来自SSR)。
Mimura等[45]用C. annuum的“California Wonder”和“LS2341”(JP187992)杂交,建立DH群体,整合了许多可重复的标记,构建了第一张具有12条染色体的C. annuum的SSR遗传图谱,与Minamiyama等[39]、Wu等[46]和Yi等[38]构建的遗传图谱基本相同。该图谱包含151个SSR、90个AFLP、10个CAPS和2个STS标记,长度为1 336 cM。因有很多基于PCR的锚定标记,其中的SSR和STS标记位置与以往报道的图谱较为一致[32,36,39],因此能够与其他辣椒遗传图谱进行比较。开发了新的CAPS和SSR引物,并对影响果实发育、生长和抗青枯病等相关性状进行QTLs定位。该图谱对C. annuum的MAS和QTLS有一定的参考价值。
Wu等[46]构建了第一张完整的遗传图谱,包含299个辣椒、番茄同源分子标记,以及263个保守的COSII标记,所有标记都覆盖在与12条染色体相对应的LGs,图谱长1 613 cM,标记间平均距离6.9 cM。该图谱基于COSII标记,因此能够推断辣椒和番茄基因组之间的亲缘关系,为茄科植物的染色体进化提供新线索。Moulin等[47]构建的C. baccatum的F2群体综合遗传图谱,由12个主要LGs和4个次要LGs组成,定位了42个SSRs、85个ISSRs 和56个RAPDs,距离为2 547.5 cM,标记间平均距离14.25 cM。为将C. annuum性状转育到C. baccatum, Minamiyama等[39]开发了62个SSR标记,其中有42个SSR定位在C. baccatum。该图谱有助于研究C. baccatum与辣椒属其他种的关系。
Sugita等[48]从6 528个克隆和13 003个cDNA/EST序列中分离出1 736个基因组SSR标记和1 344个cDNA衍生SSR标记。利用自交系杂交(K9-11×MZC-180)得到的DH系,构建了第一张以SSR标记为主的高密度种内连锁图谱(KLDH图谱),12个LGs,有597个分子标记(69个cDNA衍生的SSR,196个基因组SSR,107个先前报道的 SSR,34个 SNP/InDel,164个 AFLP,27个RAPD),遗传距离2 028 cM,标记之间的平均距离小于4 cM。与Wu等[46]构建的种间COSII图谱比较,两个图谱的遗传距离和标记分布相似,说明KL-DH图谱覆盖了辣椒的整个基因组。
Kim等[21]对C. annuum的两个品种进行了重新测序,对C. chinense野生亲缘种进行了de novo测序,认为辣椒品种CM334(C.annuumcv.)全基因组650.2 Gb(186.6×基因组覆盖率),大小为番茄的4倍。Qin等[4]报道了C.annuum的Zunla-1及其野生后代Chiltepin的全基因组序列,通过与其他茄科植物基因组相比,Chiltepin的基因组扩张了约0.3 Mya,全基因组3.48 Gb,含有34 476个蛋白质编码基因,约79%基因定位在染色体上,形成了一个高密度辣椒遗传图谱。辣椒基因组序列研究有望加速茄科植物分子标记的开发、遗传图谱的构建、候选基因的评价、经济性状的改良、进化和比较基因组学的研究。
目前,已开发出多种SNP和InDel标记,可快速构建新的辣椒连锁图谱。新一代测序技术的出现为辣椒等重要作物的高通量基因分析提供了技术支撑,如Roche/454、Solexa/Illumina和 AB SOLiD、Polonator和HeliScope等技术已被广泛应用,为了检测包括SNP和InDels在内的遗传变异,并在作物遗传学和育种中开发基于DNA的分子标记[49]。已经有一些高通量SNP基因分型平台,包括Illumina Infinium iSelect HD array(国际 HapMap 联合会[50]),Affymetrix Axiom阵列(国际HapMap联合会[50]),Douglas Array Tape(www.dougl assci entif ic.com),Fluidigm dynamic arrays[51],限制性酶测序基因型(GBS)[52]和扩增子测序[53]。Lee等[54]利用母本(C.annuum“NB1”)和父本(C.chinense“Jolokia”)杂交的F2群体,NGS重测序获得的116个SNP(HRM)标记,构建了辣椒遗传图谱(12个LGs),总距离为1 167.9 cM,可直接用于双亲不同性状的QTLS分析。
为了促进单克隆抗体MAB和辣椒基因组结构和/或组织的分析,Park等[55]利用“NuMex RNaky”(RN)(C.annuum)和 “PI159234”(CA4)(C.chinense)杂交,构建了F2群体“AC99”,利用ESTs开发SNP,将512个标记定位到12个LGs上,构建了一张高密度SNP图谱,包括214 IBP、143个 COSIS、48个 EST-SNP(ESNP)和 107个其他(84个SSR和23个RFLP)标记,图谱覆盖2 335.6 cM,两位点距离3.1(LG P8)~6.7 cM(LG P5),平均距离为4.5 cM。Li[56]利用BA3和B702(C.annuum)杂交构建了F2群体(n = 178个后代),通过对双亲的全基因组重测序,基于InDel,构建了第一张纯粹的辣椒InDe遗传图谱(BB-InDel),由12个LGs组成,包含251个InDel标记,遗传距离1 178.01 cM,标记间平均距离5.01 cM。为了加快分子标记的开发和应用,鉴定影响辣椒开花时间相关性状QTLs,Tan等[57]利用“BA3” (C.annuum)和 “YNXML”(C.frutescens)种间杂交产生的F2代,基于InDel和SSR标记,构建了一张连锁图谱,由13个LGs组成,整合了129个InDel和95个SSR标记,遗传距离1 249.77 cM,标记平均距离5.60 cM。锚定标记的遗传图谱和物理图谱的比较分析表明,该图谱几乎覆盖了整个辣椒基因组。在2号染色体上检测到一个影响开花时间的主效QTLS,命名为Nle2.2。
Lee等[58]利用C. baccatum的“Golden-aji”和“PI594137”杂交的F2(n=93),构建了一张C. baccatum的SNP遗传图谱,包含12个LGs,395个HRM标记,距离为1 056.2 cM,标记间平均距离为2.67 cM。通过与“CM334” 1.5版的物理图谱[21]比较,首次发现染色体3和染色体5、染色体3和染色体9之间相互易位,可能引起C.annuum和C.baccatum之间的生殖障碍。Mahasuk等[59]利用“Bangchang”(C.annuum) דPBC932”(C.chinense)F2群体(n = 126个体)和“PBC80”(C.baccatum)דCA1316”(C.baccatum)F2群 体(n = 146个体),创建了两个SNP基因图谱,利用高通量KASP技术,对炭疽病抗性进行QTLs定位。种间图谱(PBC932图谱)包含12个LGs和214个SNP,长824 cM,标记间平均距离为3.85 cM。种内“PBC80”图谱包含12个LGs和403个基于KASP的SNP,长1 270 cM,标记间的平均距离为3.15 cM。在“PBC932”和“PBC80”图谱上分别鉴定了2、5个与抗性炭疽病相关的QTLs。
Han等[60]利用C.annuum的“Perennia”和“Dempsey”的F7:10的120 RILs,构建了辣椒种内第一张超高密度bin图谱,含有2 578个bin,遗传长度1 372.2 cM,bin间平均距离为0.53 cM。对辣椒园艺性状进行QTLs检测,共检测到86个控制17个园艺性状的QTLs。Hulse-Kemp等[61]利用C. frutescens“Tabasco”和C. annuum“P4”杂交构建的F2群体(n = 90个体),构建了一个单倍型图谱(HapMap),5 546个标记分别定位在12个 LGs的1 361 bin中,长度1 392.3 cM。建立了辣椒高通量、高密度的SNP基因分型平台,即PepperSNP16K阵列。
Cheng等[62]利 用C.annuum“BA3” 和C.fru-tescens“YNXML”杂交的297个F2植株,构建了一个具有5 569个SNPs(3 826个遗传bin)的高密度种间遗传图谱,具有Infinium iSelect SNP阵列的Illumina(称为CapSNP15K),覆盖长度为1 628.83 cM,bin标记平均距离0.45 cM,鉴定了与果实着生方向有关的QTLS,其中一个主效QTLS命名为Up12.1,定位在LG12。根据“Zunla-1”基因组的注释,在该QTLS区域内共预测了65个蛋白质编码基因,为辣椒果实着生方向变异相关基因的分离奠定了基础。利用SLAF-seq技术,Zhu等[63]用C. chinense“740” 和C. annuum“CA1”杂交的150个F2个体,构建了一个具有种间分子高密度遗传图谱,全长1 586.78 cM,包含12个LGs,共有9 038个SLAF标记,平均距离为0.18 cM。他们还利用复合区间作图(CIM)和全基因组复合区间作图(GCIM)进行QTLS分析,确定了3个开花时期的QTLS和3个每个节点开花数QTLS。Zhang等[64]采用SLAF-seq方法,利用C.annuum品种PM702和FS871杂交的F10代的 146个RIL,获得高密度遗传连锁图谱,包含12个LGs,共有9 328个SLAF标记,遗传距离为2 009.69 cM,标记间平均距离为0.22 cM。首次在LG02上鉴定出了与辣椒第一花节紧密相关两个主效QTLs(Ffn2.1和Ffn2.2)。
辣椒的遗传图谱可以分种间[29-31,38]和种内[26,34,36,39,45,48]两种。种间群体标记多态性高,但仍受到不均等分离、种群结构安排不合理、低授粉率这些因素的影响[46],种内图谱对MAS利用有限[26]。未来,可用的遗传图谱将由多拷贝AFLPs和RAPDs以及单拷贝RFLP、SSR、SCAR、CAPS、SNP和STS分子标记组成。由于多拷贝只能在可视化凝胶上产生少数条带,单拷贝标记尽管繁琐、成本高、耗时,但比多拷贝类型标记使用将更广泛[42]。表1总结了辣椒遗传连锁图谱的简要细节。利用分布遗传图谱中LGs的标记,研究辣椒雄性不育基因的连锁标记。例如,Aulakh等[66]从遗传连锁图谱[32,38-39,67]中选择SSR标记,鉴定与NMSms10基因相关的分子标记。
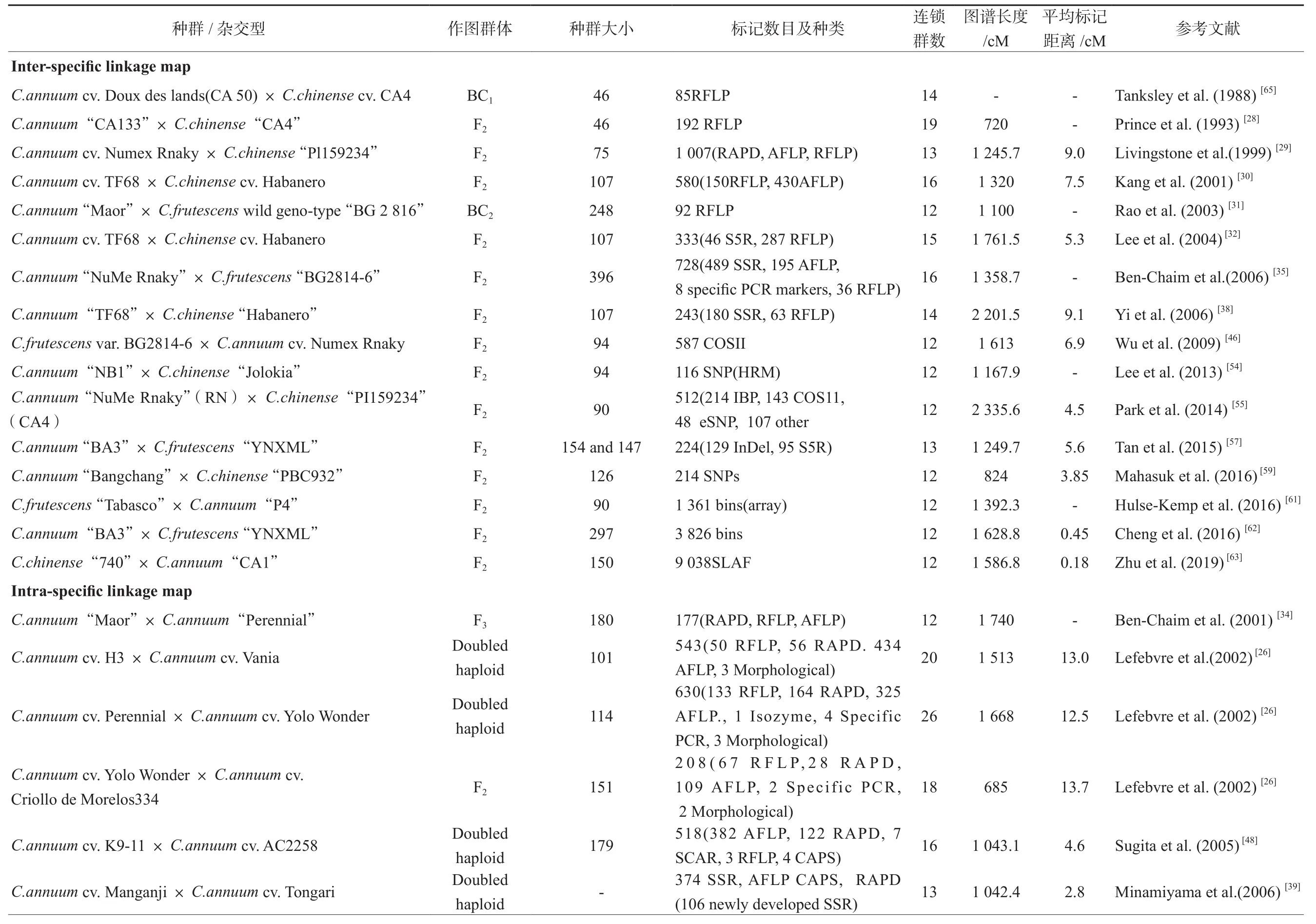
表1 辣椒遗传连锁图谱

续表1
3 辣椒核雄性不育
3.1 NMS选育和评估
通过自然突变或EMS、伽玛射线或X射线处理,选育了十几个辣椒NMS[11,68]。Martin和Crawford[8](1951)在 4526、69a和 4558等辣椒品种中发现NMS,已发现20个独立遗传的NMS基因[9]。shibriss和Frankel[68]在甜椒“All Big”中发现了一种天然的雄性不育植株(植株108-3),不同季节表现雄性不育或单性结实,于1969年选育出不育系并生产杂交种子。shibriss和Rylsky[69]将该雄性不育源命名为ms1,他们还从另一个甜椒品种“California Wonder”中获得一个稳定的雄性不育突变体,并命名为ms2。
Daskaloff[70-72]利用辐射处理诱导雄性不育突变,获得5个雄性不育基因,即ms3、ms4、ms6、ms7和ms8。用X射线处理保加利亚品种“kapia 794”的种子,在M2家系中发现一个雄性不育隐性突变体,由ms3基因控制[70]。通过对保加利亚品种“Zlaten Medal”进行γ射线辐照,获得了一个隐性NMS突变体,命名为ms8[72],该不育基因决定的雄性不育性在露地和塑料大棚中栽培表现均非常稳定[73]。
Pochard[74]在3个辣椒NMS突变体中分离出3个雄性不育基因,即ms9、ms10和ms11。Shifress[75]在甜椒品种Gambo中鉴定出一个雄性不育突变体(植株编号3633),并命名为ms12。Meshram和Narkhede[76]在辣椒品种CA 452-1中发现一个雄性不育自然突变体(ms13)。Pathak等[77]从品种“Kalyanpur selection”分离雄性不育植株,由隐性ms14基因控制。Daskalov和Poulos[12]报道了一个显性核雄性不育基因Dms,为ms5突变。中国利用两个甜椒雄性不育突变体msc1和msc2生产杂交种子[78-79]。
旁遮普农业大学将“Ludhiana”的核雄性不育基因“ms10”(原名为mc509[74])转育到法国抗多种病害品种“Punjab Lal”中,并选育了NMS系“MS12”[80],通过利用“ms12”基因培育了3个商品化杂交品种“CH-1”[81]、“H-3”[82]和“CH-27”[83]。韩 国 选 育 了“GMS1”、“GMS3”、“GMSK” 和“GMSP”等NMS系,并用于F1杂交种子的商品化生产[13, 24]。
台湾的世界蔬菜中心(WorldVeg)保存了一个源自杂交种“Novator F1”、具有ms3的NMS系,该系由匈牙利蔬菜作物研究所(VCRI)1987年提供[84],并在匈牙利生产F1种子。
3.2 标记开发和NMS基因图谱构建
已报道的20个辣椒NMS基因,只开发了几个相关分子标记。Lee等[85]建立杂交品种“Mirage”和“Fiesta”的2个F2代群体,采用AFLP-BSA技术,利用256个引物开发了1个与颜色连锁的未鉴定基因的CAPS标记,鉴定了5个多态性引物,并将一个AFLP标记Egat/Mcgg转化为CAPS标记(PmsM1-CAPS),位于ms基因2~3 cM处。采用相同的技术,通过对1 024个引物进行筛选,鉴定了 1个 AFLP标 记(514 bp)(E-AGC/M-GTG),距ms1位点3 cM,通过对MF和MS植株之间的AFLP E-AGC/M-GTG标记进行测序分析,Lee等[24]开发了一个共显性MS1-SCAR标记。在另一项研究中,Lee等[86]确定3个AFLP标记(Eagg/Mccc276、Eagc/Mctt178和 Ecag/Mtgc204) 与ms3基因紧密相连,在筛选的512个引物中,这3个与ms3等位基因共分离。对MF和MS植株之间的引物内部和侧翼区域进行测序分析,将AFLP标记(Ecag/Mtgc204)转化为GMS3-CAPS的CAPS标记。Lee等[87]鉴定了AFLP标记(E-GTA/M-ACG100),并将其转化为CAPS标记(GMSK-CAPS),与msk基因共分离,连锁距离为0 cM。然而,这些基因都没有定位到辣椒基因组上[24,85-87]。
为提高辣椒MAS的准确度,需要共分离和/或基于基因的分子标记。Jeong等[10]利用HRM分子标记,对1 118个F2代分离群体进行研究,将与ms1共分离的基因定位到C.annuum5号染色体上869.9 kb的区域。HRM是一种简单、快速、成本较低的SNP基因分型方法[88]。Kim等[21]从53个特异SNPs引物中,获得12个与ms1基因紧密连锁的HRM标记,利用基因预测分析,发现在869.9 kb序列中有11个开放阅读框(ORFs),它们与一年生辣椒参考基因组CM334编码序列(CDS)1.55相同。序列比较分析表明,基因CA05g06780,与拟南芥雄性不育系1(MS1)同源,可能是一年生辣椒的ms1的候选基因,共分离标记32187928-HRM与ms1基因紧密相连。对1 118株F2代进行分析,没有发现这种标记的重组体。利用标记32187928-HRM对48份辣椒育种材料和15个F1商品种进行有效性检验,结果表明NMS表型与标记完全相关。
Naresh等[84]以辣椒分离的F2群体(ms3基因)和甜椒稳定的F6自交系群体(msw基因)为材料,进行序列分析(GBS)和基因定位(ms3和msw)。GBS技术在利用小群体获得与单核雄性不育(ms)基因紧密连锁标记方面显示出高效性。利用GBS技术对ms3和msw基因的SNP标记进行了鉴定,在辣椒和甜椒中分别鉴定出9 713和7 453个SNP,采用BSA方法,在1号染色体(物理位置70 730 824 ~ 96 273 123)、5号染色体(物理位置32 890 811)上分别鉴定出4个与ms3基因共分离的SNPs、1个与msw基因共分离的SNPs。将SNPs转化为CAPs和dCAPS标记,对于ms3基因,开发了HPGMS2(CAPs)和HPGMS3(dCAPS)2个标记,距离3.83 cM;甜椒的dCAPS标记(SPGMS1)与msw基因完全同源。为了验证ms3基因连锁分子标记(HPGMS2和HPGMS3)的效果,对183株已知育性的F2代进行检测,这两个标记均能将Ms3ms3(杂合子MF)、Ms3Ms3(纯合子显性MF)和ms3ms3(纯合子隐性mS)基因型分开。与msw基因相关的标记SPGMS1与不育性共同分离,能将杂合(Mswmsw)MF植株与纯合隐性(mswmsw)MS植株区分开来。这些标记能在苗期对MS和MF单株进行鉴定,用于MAS和杂交种子生产。作者还认为,msw(32 187 928)的分子标记位于Jeong等[10]报道的ms1基因869.9 kb(31 516 016 ~ 32 385 930)区域内,msw和ms1很可能是同一基因(等位基因);基因测试将有助于确认msw和ms1之间关系。
Bartoszewski等[89]利用不育系 320×Elf(MF)杂交,构建甜椒F2群体,利用RAPD-BSA技术,鉴定了7个与ms8位点相关的标记。RAPD引物,N16-800、P15-530、V17-320、W06-520和Z05-760在偶合期,Q07-1100、V01-1000在分离期与ms8基因连锁。将4个RAPD标记转化SCAR-P2、SCAR-V17、SCAR-N2和 SCAR-V01标记,SCAR-P2和RAPD-Z05-760最接近ms8位点(4.6 cM),SCAR-V17和 RAPD-W06-520分别位于6.8和7.4 cM处,SCAR-N2、RAPD Q07-1100和SCAR-V01分别在16.1、17.5和18.1 cM处,所有标记均位于ms8位点的同一侧,LOD = 3。此外,他们还鉴定了2个与ms8基因相关的COSⅡ/CAPS标记。利用Lefebvre等[26]、Wu等[46]的辣椒参考图谱进行比较,表明ms8位点位于辣椒4号染色体的下臂。但与ms8基因座紧密相连的标记仍然缺乏。Lee等[90]开发了一个HRM标记GMSE,该标记与甜椒中未知的NMS位点连锁,定位在AC99图谱[42]的5号染色体上,距标记TG23 、TG363距离为 8 cM、6 cM。TG363是Lee等[42]开发的AC99和SNU7图谱中的一个RFLP通用标记。
在基因组重测序的基础上,Cheng等[91]确定了一个强候选基因,即Capana02g002096,位于C.annuum的msc-1位点,命名为Msc-1,该基因为编码拟南芥DYSFUNCTIONAL TAPETUM1(DYT1)的同源基因,有7-bp的缺失,是一个bHLH转录因子,参与早期的绒毡层发育,控制雄性不育。基于7-bp缺失,开发了一个indel标记——indel-12。为了确认标记Indel-12和MS性状之间的共分离,建立了两个NILs群体,一个510株F2群体(17Q3626),一个46个辣椒自交系(均为MF)群体。基因鉴定结果表明,在所有群体中,标记Indel-12与msc1位点共分离,从遗传学上推测,msc-1基因位点可能在C.annuum辣椒ms1基因的上游。在已发现的20个NMS基因中,仅韩国利用msk基因进行辣椒育种。利用SNP标记,Jeong等[92]绘制了msk位点附近精细图谱,通过对807株F2群体中MF(MskMsk)和Ms(mskmsk)全基因组序列,鉴定了SNP标记,开发与定位了15个HRM标记,msk基因定位到10号染色体上的145 kb基因组区域。
Aulakh等[65]定位了C.annuum雄性不育基因ms10。采用BSA法检测了F2群体,筛选了558对SSR引物,发现2个标记AVRDC-PP12和AVRDC_MD997,与ms10位点的连锁距离分别为7.2 cM和20.8 cM,定位在1号染色体175 694 513 ~175 694 644、175 438 699 ~ 175 438 872。但辣椒属中其他种NMS基因相关的标记仍然缺乏。表2列出了与NMS基因相关的可用标记。
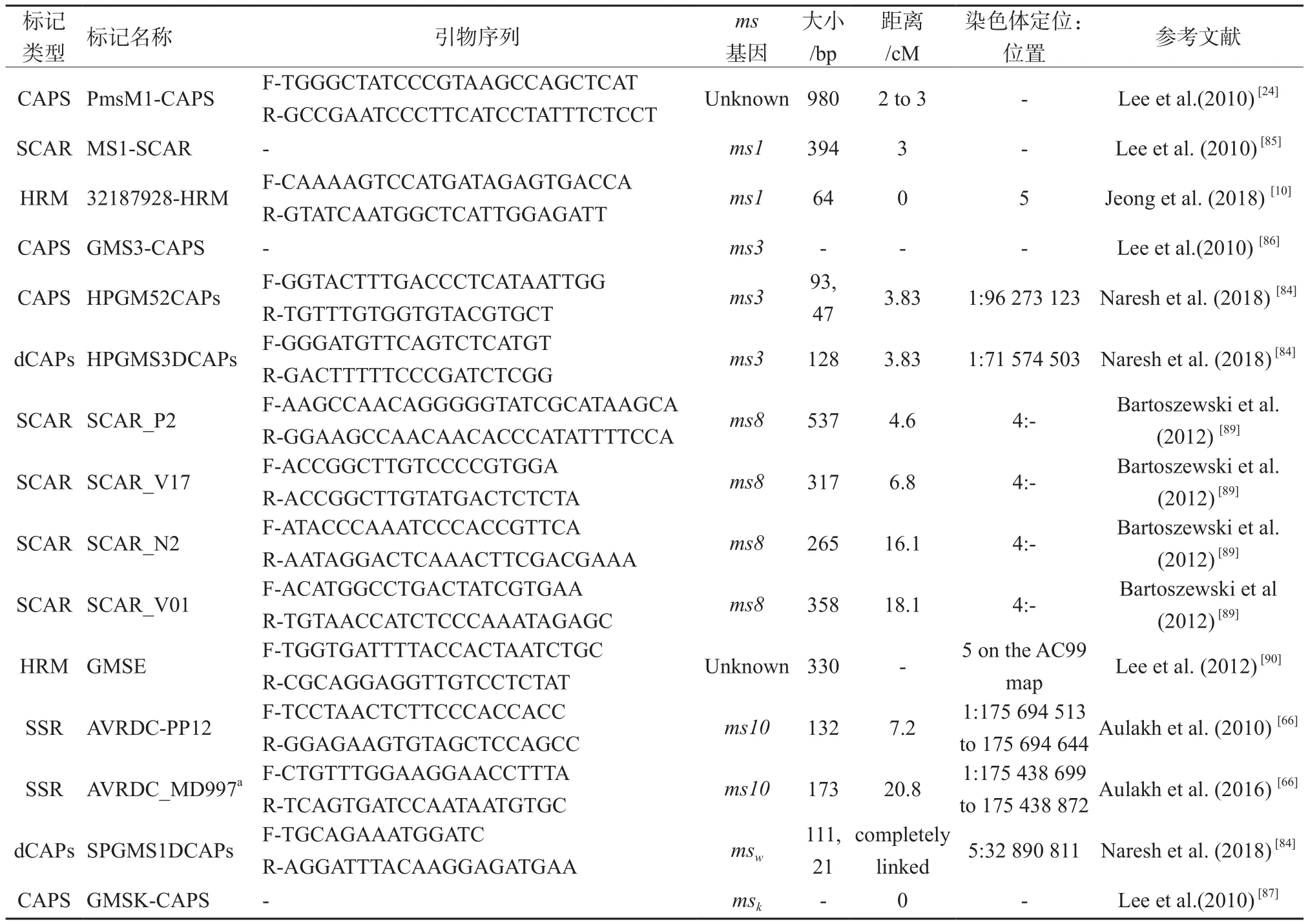
表2 与NMS基因相关的可用标记
这些标记在杂种优势育种中有着广泛的应用前景。由于不育性由隐性基因控制,用常规回交方法选育新的NMS系繁琐、费时[24]。共显性标记能区分MF植物是杂合子还是纯合子[19],可从基因型水平上进行选择,无需自花授粉再回交或杂交,从而节省时间和资源。使用MAS选育新的NMS系,可节省50%时间,繁殖种群的数量可以大大减少。利用标记辅助技术,Aulakh等[93]将NMS系“MS-12”的ms10基因转育到另外优良辣椒自交系中。对“MS-12×VR-16”、“MS-12×S-217621”、“MS-12×Selection Dev” 和“MS-12×DCL-524” 等 4个回交后代(BC2F2)进行筛选,开发了SSR标记“AVRDC-PP12”。结果表明,在BC2F2的前3个杂交后代中,该标记与ms10基因共分离,重组频率分别为3.22%、4.16%和3.27%,交换值小于5%,因此该标记可作为ms10基因分子辅助选择标记,用于不育基因的转育。
综上所述,与辣椒ms基因紧密连锁的分子标记有:(1)ms1相关标记2个,MS1-SCAR标记[24]和HRM标记(32187928-HRM)[10]。(2)ms3相关标记 3 个,GMS3-CAPS 标记[86]、HPGMS2(CAPS)和 HPGMS3(dCAPS)标记[84]。(3)ms8相关标记2个,SCAR-P2和RAPD Z05-760标记[89]。(4)1个msw基因连锁的dCAPS标记(SPGMS1)[84]。(5)1个与msk基因连锁的GMSK-CAPS标记[87]。(6)1个与msc1基因连锁的indel-12标记[91]。(7)1个与未知基因连锁的GMSE HRM标记[90]。这些标记与不育基因连锁共分离,在不育基因转育和辣椒杂种优势育种计划方面具有潜在用途。
为了提高MS植物的比例,Shifriss和Pilowsky[94]杂交了两个携带ms1和ms2基因的等基因系,期望双不育基因MS×MF后代在单基因分离中产生75%的MS植株,而不是50%。通过常规育种,聚合多个基因是非常困难的。MAS能有效聚合多个不育基因,提高选育NMS系效率。利用共显性标记[24,66,85-87,89]在苗期区分MS和MF植物,因此可以确保种子生产区的母本100%纯度。
4 辣椒胞质雄性不育
4.1 不育系的选育与评价
一般甜椒具有保持基因(rf)[11,22-23,95-97],通过将甜椒的rf基因转育到细胞质不育(S)材料中选育新的CMS系[95]。shifress和Frankel[95]将23个小果辣椒品系与3个具有rf基因的甜椒品系杂交,寻找细胞质不育新来源。两个群体(1005株和1006株)的分离模型表明,可育对不育为显性,不育受单隐性基因控制。
Madhavi Reddy等[98]选 育 了 MS-1、MS-2、MS-3和MS-4等4个不育性稳定的CMS及其保持系。Pákozdi等[99]报道不育系 202、203、204和205在5月、6-7月和8-9月3个生长季节中没有形成有活力或功能的花粉。Yazawa等[100]成功地培育出了2个不育性稳定辣椒CMS“P-MS”和“Murasaki-MS”。Liu等[101]在台湾世界蔬菜中心育成了3个花粉活性差的不育系“CCA4759”、“CCA4757”和“CCA5273”,这些不育系在低温(< 23.8℃/14.3℃(昼/夜))下不结籽。世界蔬菜中心的Gniffke等[102]对5个不育系进行了评价,发现“CCA7234”和“CCA7235”2个不育系在冬季不育性表现稳定。Gong等[103]通过种间性杂交和诱导突变相结合的方法,选育了几个辣椒CMS系、保持系和恢复系。
为了评估Peterson的不育源的利用价值,用270个辣椒自交系与该不育源杂交,观察后代的育性分离,Yu[96]发现,152个自交系可作保持系((N)rfrf),66个自交系可作恢复系((N/S)RfRf),其余的归类为不稳定系。经自花授粉和选择,能选育出稳定的保持系和恢复系[97],不稳定系可能是不育修饰基因的杂合系,其后代对年份或季节变化有反应。
Kim等[104]评估了辣椒热敏感胞质雄性不育系(Milyang-A)。他们观察到,该系在15℃以上不育,但当夜间温度降到13℃以下时,它的繁殖能力得到恢复。在另一项研究中,200A×206B、201A×200B、201A×206B、203A×200B、206A×200B、206A×201B的自交结实率为零,6个辣椒杂种F1没有正常花粉,表明杂种F1不育[105]。Suryawanshi、Kulkarni和 Baig[106]报告说,辣椒不育系“CMS-a(0246)”植株在不同地点都不结果。湖南省农业科学院蔬菜研究所培育的辣椒不育系8214A,具有100%的雄性不育性,良好的配合力、农艺性状和经济性状,常用来进行辣椒杂交制种。
利用Gniffke等[102]选育的S-细胞质不育源,培育具有不同遗传背景的优良不育系,经过6代回交和筛选,选育了10个温稳不育系,分别是“CMS4611A”、“CMS4614A”、“CMS4622A”、“CMS4624A”、“CMS4626A”、“CMS46213A”、“CMS463D2A”、“CMS463D13A”、“CMS463D14A”和“CMS463L5A”[107]。采用SSR标记分析,对CMS的 3个 A 系“CMS4611A”、“CMS4626A”和“CMS463D13A”及其B系进行评估,筛选了覆盖辣椒全基因组的120个SSR标记(每个连锁群10个),这些SSR标记来自前人构建的连锁图谱[32,38-39,67]。结果表明,经过5个回交周期(BC5F1)的选择,3个温稳型CMS的A系基因组恢复率达96%以上。这些不育系遗传稳定,正用于杂交育种计划[108]。
4.2 CMS基因的标记开发和定位
从Peterson[14]报道胞质雄性不育系PI 164835后,科技人员开展了一系列分子机理研究,以揭示CMS不育的原因。利用Southern blot-RFLP分析,认为线粒体中的coxII和atp6-2基因组区域可能与C.annuum的不育有关[109]。Ψatp6-2在atp6-2的3'端的编码区被截断[110],Northern blot表明,不育系和恢复系的mRNA带型存在差异,表明atp6基因是辣椒不育的候选基因之一。Kim等[111]在不育系的coxII基因(与coxII共转录)的3'端,发现了一个嵌合不育相关线粒体新开放阅读框(orf456),不育系表达约17-kDa,恢复系中该蛋白的表达水平很低。在拟南芥线粒体表达,转化群体花粉量和雄性不育表型均存在缺陷,orf456蛋白功能得到证实[111]。说明新发现的orf456是一个强不育候选基因,可以用来鉴定辣椒CMS的不育表型。另一项研究表明,细胞色素C氧化酶活性和F1F0ATPase酶的异常可能导致了不育系的花药败育。Ψatp6-2基因可能导致F1F0ATPase功能紊乱,orf507控制着细胞色素C氧化酶的活性,从而导致不育系花药败育[112]。
Kim等[113]利用辣椒Milyang-A(S-细胞质)和Milyang-B(N-细胞质)的近等基因系,在侧翼序列中获得了2个不育基因标记,SCARatp6607标记,SCARcoxII708标记,并在18个辣椒CMS对标记准确性和可靠性进行验证。SCARatp6标记为607 bp,来源不育系,在保持系中未扩增出该片段。coxII-SCAR为S细胞质的特异PCR片段(708 bp)。Jo等[114]用辣椒CMS系“FS4401”和自交系“Jeju”,开发了一个新的不育标记accD-U,用accD-U标记筛选CMS及保持系,仅在不育系中扩增出1 082 bp片段,保持系中没有。因此认为选育MS系时,比Kim等[113]报道的标记更有效、更可靠、更有用。Gulyas等[115]检测到已报道的orf456基因的转录本发生了改变,并命名为orf507,并对不育系植株的线粒体进行了测序。发现当PCR循环次数超过35次[116]时,orf456和atp6标记在少数辣椒保持系(B系)中被轻微扩增,这可能会影响胞质雄性不育鉴定的可靠性。利用线粒体基因组序列相关SRAP标记,Ji等[117]2014年开发了一个新的鉴定一年生辣椒胞质雄性不育的特异SCAR130标记,在不育系中产生130-bp的PCR产物,在保持系中产生140-bp的片段,没有产生轻微扩增。结果比已报道的标记(orf507、atp6和accD-U)更可靠。SCAR130位于烟草线粒体基因组trnE和trnS之间,连锁基因在不育系HW203A花粉败育期(四分体期)的表达显著高于保持系。因此推测除orf507和atp6外,CMS还可能存在其他调节因子。与辣椒CMS相关基因相关的分子标记如表3所示。
多数研究表明,CMS育性恢复是由一个可遗传的“Rf”显性基因决定,也不排除互补基因[124]、修饰基因[116,125]、小基因或多基因[126]、第三个单倍型的Rf位点[17]和环境因素[15,124]参与了育性恢复。Min等[16]的后续研究排除了第三个单倍型的Rf基因。
在平面作画转移至在立体的器型上作画时,是需要对所展现的事物造型的进行相应的处理和调整的,对整体的布局也窑技能型重新的规划,不然,画面的最终效果就会不如人意。因此,在将花鸟题材的作品移至器物之上的时候,我们需要对画面整体进行布局,这与国画的整体格局的布置有着异曲同工之妙。然后,再对题材的造型进行一定的处理,最终才能以最好的方式呈现出画面的美感。
Zhang等[23]应用RAPD-BSA方法,首次报道与辣椒Rf基因连锁的RAPD分子标记。用21号牛角椒-A(rfrf)×湘潭晚(RfRf)的F2代群体,发现2个与Rf基因相关的RAPD标记,标记OP131400与该基因紧密连锁,遗传距离为0.37 cM,第二个标记OW19800位于对侧,遗传距离为8.12 cM,LOD值分别为46.81和23.38。OP131400为后续研究中最常用的Rf连锁标记。Kim等[120]把OP131400RAPD转化为OP13-CAPS, Min等[17]根据OP13-CAPS生成CaRf-M1标记。Kim等[110]和Min等[17]发现OP13-CAPS距离恢复基因位点1.1 cM,但Min等[16]显示其距离为0.4 cM,类似于Zhang等[23]最初报告的0.37 cM。
Gulyas[119]开发了一个CRF3S1S SCAR标记,在F3和F4的群体中,距Rf位点分别为5.3和4.8 cM。Min等[16]将CRF3S1S SCAR标记转换为CAPS标记,并确认该标记比Gulyas等[115]、Kim等[120]报道中检测到的8个AFLP标记(AFRF1到AFRF8)更接近恢复基因位点,距离1.5 cM,后成功将1个(AFRF8)转换为一个名为AFRF8 CAPS的CAPS标记。此外,利用TS502(rfrf)×HK6T(RfRf)的138个F2植株,构建了一个全长54.1 cM的连锁图谱(LOD = 4.0), 包含1个AFRF8 CAPS、1个OP131400RAPD 和 7个AFLP 标记。AFRF8 CAPS标记位于恢复基因位点1.8 cM处,它和OP131400RAPD标记位于基因的同一侧[17]。Zhang等[23]研究认为,Rf位点的侧翼是RAPD标记OP131400,距离为1.1 cM,与本研究不同,可能是由于使用两张图谱所致,他们还试图在辣椒“SNU2”图谱[32]上定位AFRF8 CAPS和OP131400RAPD标记,但没有获得相应结果。AFRF8 CAPS衍生的CaRf-FL-M2标记位于距离CaRf-M1 标记 0.1 cM[17]。
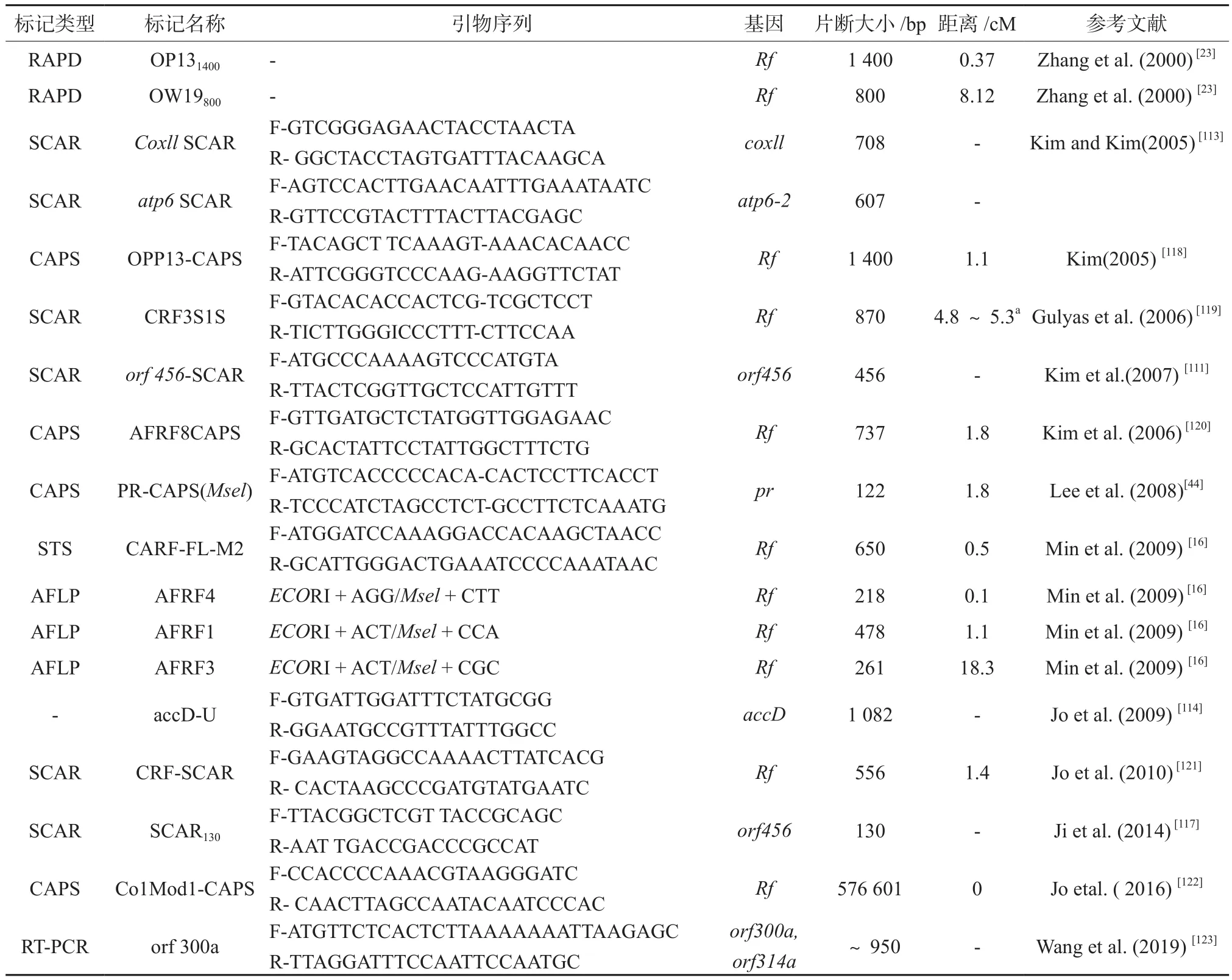
表3 与辣椒CMS相关基因连锁的分子标记特征
Min等[116]用辣椒杂交品种“Buja”和“Tamna”的F2群体,构建了Rf-BSA、rf-BSA和4个重组子(1个MS和3个MF)池,用于AFLP标记分析,鉴定并筛选出3个Rf连锁分子标记AFRF1、AFRF3和AFRF4,构建了连锁图谱。标记AFRF4与Rf基因型和4个重组子共分离,与Rf位点的最短遗传距离0.1 cM,位于Rf位点的对侧。
Jo等[121]认为已克隆的Rf基因可作为开发辣椒Rf候选基因连锁标记。用已克隆的矮牵牛Rf作为候选基因,利用3个BAC库,开发了BAC13T7 SCAR(1.4 cM)、BAC17T7 SCAR(3.2 cM) 和PepPR1-as-PCR(14cM)等3个Rf连锁标记,矮牵牛同源基因位于辣椒Rf位点附近。在已开发标记[120,125]基础上,Gulyas等[119]开发出适用性更广泛的CRF-SCAR标记,遗传距离更近(1.4 cM)。与Lee等[125]比较,CRF-SCAR和OPP13-CAPS被定位在基因位点的对侧。并将已开发和新开发的标记定位在辣椒AC99图谱的6号染色体上端[42]。
为了解主效Rf基因、次效基因和温度等环境因素的作用,Wang等[126]确定了恢复能力的数量性状位点(QTLS)[42],利用综合遗传图谱[26]中的标记,将1个恢复主效QTLS定位在6号染色体上端末,占表型变异的20%~69%;4个次要QTLS定位在其他染色体(5号、2号,连锁群PY3和PY1)上,占表型变异的7%~17%。Jo等[121]研究表明,OPP13-CAPS和其他Rf连锁标记在同一区域(6号染色体的上端末),因此认为Wang等[126]报道的主效Rf和Rf-QTLS可能是同一基因。尽管开展了很多研究,Rf基因的实际图谱位置还没有确定。
Lee等[18]确定了CMS中影响恢复能力的相关“pr”(部分恢复)位点。主要“Rf”基因与隐性的“pr”基因共存,表现部分恢复。这种植株有正常的花药,同时产生正常和败育花粉,但多数花粉开裂后粘在花药壁上,导致果实种子少,坐果率低。Lee等[125]利用768个AFLP标记进行AFLP-BSA分析,鉴定出8个Pr-连锁AFLP引物。利用AFLP标记,对GD×HF3-SC1的87株F2后代进行筛选,在8个AFPL标记中,有2个标记E-AGC/M-GCA122和E-TCT/M-CCG116与位点最近,遗传距离估计为1.8 cM。根据AFLP片段序列和侧翼区域的序列,将E-AGC/M-GCA122转化为 PR-CAPS标记。利用1个与pr-连锁标记(PR-CAPS)[125]和3个Rf-连锁标记(OPP13-CAPS[23,118]、AFRF8-CAPS[120]和CRF-SCAR[32,119]),对韩国商品辣椒“Buja”(S,Rf/rf)的205株F2进行分子生物学分析,确定部分恢复“pr”与育性恢复“Rf”基因位点的连锁关系,发现这四个标记紧密相连,表明pr基因与Rf位点相关或是Rf位点的第三等位基因,其显性关系为Rf > pr > rf[18]。
大量的种质资源评价结果表明,恢复系来源有限,大部分甜椒和相当一部分辣椒缺乏Rf基因[22-23],制约了杂交育种中恢复系的选育。辣椒Rf及其相关基因的分子生物学研究有助于辣椒Rf/rf基因的快速筛选,无需评价杂交后代育性,从而加速rf基因、Rf基因分别在保持系、恢复系的转育。在许多已开发的标记中,OP131400RAPD[23]、CaRf-M1[17]和AFRF4[16]与Rf紧密连锁,位于Rf位点的对侧,这些标记将在MAS发挥重要作用。在MAB育种中,运用Rf位点连锁的SCAR标记CRF-S870(CRF3S1S)[119],将Rf等位基因导入甜椒新基因型[127]。在另一项研究中,用10个辣椒、10个甜椒CMS系及其保持系与5个辣椒、5个甜椒恢复系,Yeh等[128]验证了5个CMS(S细胞质)不育性(atp6-SCAR607,Ψatp6-2875,coxIISCAR708,orf456,SCAR130/140)和 1 个Rf位点恢复性(CRF-S870),在5个不育性的标记中,共显性SCAR130/140[117]是检测细胞质类型(S vs.N)最可靠和可重复的标记,该标记与pr基因相关[125],将有助于选育C系,通过消除pr等位基因的影响,完全恢复不育系的育性。
Mulyantoro等[129]利用CMS相关标记,将甜椒NMS转化为CMS。2个CMS特异不育性标记atp6SCAR和coxIISCAR[113],2个Rf连锁标记CRF-CAPS[119]和 BAC13T7-CAPS[121],Mulyantoro等[130]首次将甜椒品种“GC3”NMS成功转化为CMS。辣椒具有S细胞质和Rf等位基因,这些发现将有助于提高甜椒杂交制种产量。
Wang等[123]利用Illumina PE和PacBio技术,对辣椒CMS系“138A”及其保持系“138B”的线粒体基因组进行测序和组装。CMS“138A”的线粒体基因组504 210 bp,比“138B”短8 618 bp;在“138A”、“138B”中鉴定出大于100个氨基酸的ORFs分别为214个、215个。138A与138B线粒体基因组结构有较大差异,存在重组和重排事件。通过比较两个线粒体基因组,选择了几个可能与不育有关的ORF(orf300a、orf314a、orf262a、orf292a、orf157a、orf115b), 其 中orf300a和orf314a是138A中控制不育的强候选基因,并开发了一个与不育性状共分离新的标记(orf300a)。
5 雄性不育相关基因的克隆及不育系的转录组/蛋白质组分析
在辣椒中已经发现了一些与NMS基因相关的分子标记。但迄今为止,辣椒中NMS的分子机制尚不清楚,关于辣椒中NMS相关基因的克隆、鉴定和功能分析的报道很少。
利用一年生辣椒开展核雄性不育系“114AB”研究,发现一个花药特异性脂质转移蛋白(LTP)基因CaMF2影响花粉发育[132]。采用cDNA-AFLP,首次鉴定和描述了1个新的雄性不育相关基因Camf1,编码乙二醛氧化酶相关蛋白[133]。表达分析表明,CaMF2和Camf1基因只在可育株已发育的花药中表达,而在不育株中不表达,说明这些基因可能在花粉发育中起重要作用。CaMF2和Camf1的抑制或缺失可能导致辣椒不育。Camf1的长度为1 854 bp,不包含1 707 bp ORF的内含子。Chen等[132-133]有关CaMF2和Camf1的研究为理解辣椒中NMS的分子机制提供了一种途径。此后,使用相同的NMS系(114AB),Hao等[134]克隆并鉴定了一个新的乙酸异戊酯水解酯酶(IAH1)基因CaMF3,该基因仅在发育后期的花蕾和可育辣椒株的开放花中表达,表明CaMF3参与了后期花粉发育,在花粉成熟和萌发过程中起一定作用。此外,对CaMF3的研究有助于进一步了解花药发育的分子机制以及IAH1基因家族在辣椒花药或花粉发育中的作用。Chen等[135]利用RNA-Seq技术对辣椒核雄性不育株和可育株进行全基因组转录分析,用于确定花粉发育和成熟的候选基因。为了进一步研究与花粉发育相关的基因,Hao等[136]在辣椒中克隆并鉴定了一个新的花药特异基因CaMF4。
细胞质雄性不育和育性恢复的分子机制仍不清楚。Liu等[137]利用NGS技术,对辣椒CMS的“121A”和 “121C”之间的差异进行转录组测序和从头分析,找出与MS相关的关键基因和途径。以恢复系为参考,鉴定出4 326个上调单基因(在不育系“121A”中高表达)和7 061个下调单基因(在恢复系“121C”中高表达)。许多差异表达的单基因代表了一组与花粉形成或败育相关的潜在候选基因。在进一步的富集分析之后,确定了三条富集途径,覆盖了差异最丰富的单基因。
为了从蛋白质组水平上揭示辣椒细胞质雄性不育的分子机制,Zhang等[138]利用透射电镜(TEM),研究不育系FS1030A及其保持系FS1030B在小孢子母细胞败育过程中的花药蛋白质组。与细胞质雄性不育或导致细胞质雄性不育相关的蛋白质通常在MS系中表现出较低或较高的表达。在检测到的全部蛋白质点中,有13个蛋白(7个上调,6个下调)在“FS1030A”和“FS1030B”中有差异表达。此外,使用国家生物技术信息中心(NCBI)的绿色植物和辣椒数据库,13个蛋白点中有12个被鉴定为9个单独的蛋白,天冬氨酸蛋白酶的过度表达导致了正常的程序性细胞死亡(PCD),导致FS1030A不育。这是第一次用蛋白质组学方法研究辣椒CMS。Guo等[139]利用“无标记”高通量蛋白质组学方法,对辣椒CMS的不育系及其保持系的表达谱进行比较,以揭示辣椒CMS的潜在机制。鉴定和定量了324种差异丰富蛋白质(DAPS),其中47种和140种分别在A系中向上和向下积累。此外,在CMS A系和B系中分别特异性地积累了75种和62种蛋白质。花粉外壁形成、三羧酸循环(TCA)、线粒体电子传递链(mtETC)、丙酮酸代谢过程和氧化应激反应中所涉及的蛋白质种类在A和B系之间存在差异积累,表明它们在辣椒花粉败育的调控中具有潜在的作用。
为了解雄性不育的机制,Qiu等[140]采用RNA-Seq方法对不育系“8214A”及其保持系“8214B”进行比较转录组分析。共有1 355个基因被确定有差异表达,“8214A”和“8214B”在减数分裂期间花药中相关基因表达水平有差异,有424个和931个上调和下调基因。20个泛素连接酶和3个细胞周期相关基因的表达水平在减数分裂过程中被强烈下调,甲基转移酶基因在“8214A”花药中表达上调,可能导致“8214A”花粉母细胞在减数分裂中程序性死亡,结果有助于揭示CMS辣椒花粉败育的分子机制。
Li等[141]克隆辣椒线粒体ATP合成酶6kDa亚基(MtATP6),认为“Milyang-A”细胞质雄性不育可能是由于MtATP6活性降低引起的,因为较少的ATP含量可能影响了细胞质雄性不育植株的花粉发育,这可能是由核编码的MtATP6和线粒体Orf507之间的相互作用引起的。Livingstone等[29]对 NuMex RNaky(RNaky)(C.annuum)和PI159234(CA4)(C.chinense)杂交的 70株 F2进行研究,将MtATP6定位在4号染色体NP1234和TG1324标记之间区域。
6 杂交种子遗传纯度检测
明确品种的一致性、特异性和稳定性,对保护育种人员的权益和有效控制种子质量具有重要意义。这些控制措施旨在验证指定的杂交已经发生,母本自花授粉或同胞授粉数量符合必要的纯度要求,种子活力符合标准[142]。
雄性不育系已在辣椒杂交制种应用。在印度等一些国家,NMS已在辣椒杂交种子生产上广泛应用[81-83]。正如之前讨论过,NMS有育性分离,需要尽早拔除可育植株。CMS受环境因素影响,特别是低温影响[14-15,124]。这两种情况都会导致杂交种子受亲本自花或同胞花授粉的影响。因此,为了保证杂交种子的纯度,需要开发高通量评价系统。
种子纯度一般通过田间生长遗传特性(GoT)或分子标记来判断,后代表现隐性性状,一般认为是自花授粉的结果。Jang等[143]、Mongkolporn等[144]认为 GoT鉴定在时间、成本和复杂性方面有许多缺点。由于品种间和品种内的遗传多样性有限,品种内的形态标记很难识别[142]。与此相反,分子标记数量众多,且与环境效应无关。
利用RAPD、RFLP、SCAR、SNP和STS等多种PCR标记对杂交种进行亲本鉴定和遗传纯度检测。Jang等[143]考虑了6个标准:准确度、再现性、快速性、成本效率、简单性和实用性,以解决杂交种纯度测试的现有缺陷。尽管RFLP标记具有更好的多态性和稳定性[145],但由于其高成本和复杂性,在常规商业检测中具有局限性[146]。在众多用于品种鉴定和纯度检测的分子标记中,由于RAPD具有显性遗传,父本特异性标记有助于杂种纯度检测,同时具有成本低、使用方便等优点,RAPD是成为最常用的一种方法。
Ballester和de Vicente[142]首次报道了用RAPD标记检测辣椒杂交种子纯度。筛选100对引物,其中“OPV-06”为检测5个杂交种纯度的最有效引物。Choe等[147]同时使用RAPD标记和磷酸葡萄糖变位酶(PGM)同工酶分析进行亲本鉴定和种子纯度检测,认为用高纯度的亲本作参考,通过比较RAPD,可以进行F1纯度检测。Jang等[143]开发了2个RAPD标记,UBC336700作为母本特异性标记,UBC147850作为父本特异性标记,并将其转化为基于PCR的SCAR标记。在盲试中,RAPD和SCAR标记能够可靠地检测出非本品种。Kumar等[22]利用Rf-连锁 RAPD标记(OW 19 800和OP 131 400)对“CCA 4261”דPusa Jwala”进行杂交纯度检测,这两个标记都是父本的特异性标记,很容易检测自交后代。
SSR标记具有高度变异性、共显性遗传、多等位性、重复性和良好的基因组覆盖率。一个单一的多态性标记足以检测来自其亲本的污染,两个或多个标记检测杂交种子的外源污染更为有效[148]。然而,目前SSR标记尚未用于辣椒种子纯度的检测。
7 总结
NMS系和CMS系均已用于辣椒杂交育种。为了加强杂种优势育种,MAS对促进亲本选育具有重要意义。在所鉴定的20个NMS基因中,只有ms1、ms3、ms8、ms10、msk、msc-1和一个未鉴定的基因的标记被开发出来。除ms1、ms3、ms8、ms10和未鉴定的基因外,其他基因位点尚不清楚。最近报道的辣椒连锁图谱和全基因组测序将加速基因的标记和定位。这些紧密连锁的标记将有助于通过MAS选育NMS系。线粒体基因atp6是辣椒细胞质雄性不育的候选基因。在辣椒CMS相关基因中,Rf是研究最多的一个。该基因已定位在第6号染色体上,但尚未克隆。基因精细作图将定位重叠或更接近位点的标记,有利于辣椒种质资源的快速筛选,加速rf基因、Rf基因分别在保持系、恢复系中的转育。甜椒Rf基因稀缺,CMS相关标记有助于甜椒优良的NMS转育为CMS系,这将提高杂交育种效率和杂交种子纯度。
