基于简化基因组数据开发岭南青冈微卫星引物
2020-07-13姜小龙徐刚标
宁 馨 姜小龙 邓 敏 徐刚标
(1.中南林业科技大学林木遗传育种实验室,长沙 410004;2.中国科学院上海辰山植物科学研究中心,上海辰山植物园,上海 201602)
微卫星又称为短串联重复序列(short tandem repeats,STRs)或简单重复序列(simple sequence repeat,SSR),通常由1~6个核苷酸的串联重复片段构成,一般重复几次到几十次[1]。跟核苷酸的遗传多样性及突变率相比,微卫星具有更高的多态性及突变速率。因此,自20世纪90年代开始,微卫星标记就一直被广泛用于法医、亲子鉴定、保护遗传学、谱系地理学及群体遗传学中[2~3]。不过由于微卫星的侧翼序列在种间也可能存在变异,因此,针对某一物种设计的引物通常只适用于这一物种及其近源种。随着近几年新一代测序技术的快速发展,基于高通量测序技术可在短时期内以较低成本获得大量SNP(Single nucleotide polymorphism)[4]。例如,近几年出现的简化基因组测序技术(restriction-site associated DNA sequence,RAD-seq)现已在群体遗传学、保护生物学及谱系地理学中开始广泛使用[5]。目前微卫星在保护生物学及群体遗传学中还具有一定的优势。首先,微卫星标记能有效地扩增DNA降解严重的样本。虽然RAD-seq能以较低的成本得到大量的直系同源SNPs,但是其对提取DNA质量的要求较高,很多长期硅胶干燥保存的分子样和标本上收集的样本上提取的DNA及古DNA往往不能满足二代测序的建库要求;其次,RAD-seq中所得到SNPs的错误率跟DNA降解程度呈现正相关[6];再次,微卫星的成本低廉。尽管利用微卫星标记得到的变异位点有限,但对于较少个体的濒危物种群体遗传学研究,微卫星标记仍是一个有效、快捷的手段。并且微卫星数据易于分析。使用普通的个人电脑就可以对其进行数据分析和处理,而RAD-seq产生的数据量较大,通常需要服务器或者工作站进行数据分析,并需要有熟练的数据分析技能。
经典微卫星引物开发的方法有文库法、磁珠富集法和省略筛库法。文库法是先用超声波或酶切方法将DNA切成小片段后建库,然后用含有SSR的探针与文库杂交,筛选出和探针有较强信号的克隆,此方法开发引物所得到的SSR阳性克隆比率低,不过操作简单、容易掌握。富集法通过建立和筛选微卫星富集文库,能极大地提高文库法中SSR克隆得率低的问题。例如,磁珠富集法用生物素标记重复序列特异探针并与基因组DNA酶切片段杂交,再用包被链亲和素的磁珠对生物素进行磁力吸附,从而完成重复序列目标片段的富集。省略筛库法使用5′端带7个简并碱基的SSR引物(5′-KKVRVRV(CT)6-3′)对基因组SSR区域进行PCR富集后进行测序[7]。此方法不需要筛库去识别含SSR的克隆,可以进一步提高有多态位点SSR的得率。近年来,随着测序技术的快速发展,高通量测序技术被广泛用来开发微卫星引物。例如,使用物种转录组序列[8~10]、基因组序列[11]和简化基因组序列[12~13]来进行开发。这些方法直接利用测序序列设计SSR引物,简化了实验流程,并可得到更多的微卫星位点。先前使用简化基因组序列开发SSR引物都是对双端测序数据(2×150 bp)拼接后获得较长的序列,然后再开发引物[14]。SSR扩增片段长度通常多为100 bp,因此,是否能直接使用简化基因组测序中150 bp的R1端序列直接设计引物呢?如果可行,则能进一步简化引物设计的流程。
本次研究我们基于前期获得的岭南青冈(QuercuschampioniiBenth)RAD-seq数据的R1端序列,进行SSR引物的开发,并作验证。岭南青冈是中国南方及中南半岛亚热带中山云雾林的代表种,主要分布于海拔100~1 700 m的森林[15]。本研究旨在证明基于简化基因组测序获得的R1端序列(150 bp)是否能够开发出足够的SSR引物用于群体遗传分析。
1 材料与方法
1.1 样品来源
本研究所用的4个样本来自于Jiang等文章中4个不同群体的岭南青冈简化基因组数据,分别为广东省龙门县南昆山、广州省佛冈县观音山、广西省宁明县公母山和广东省罗博县象头山[16]。
1.2 数据过滤与引物设计
数据的过滤与引物设计按照如下流程进行:
(1)使用pyRAD[17]对序列进行质控过滤,去除低质量的reads。使用SciRoKo v3.4[18]提取包含有微卫星重复序列的reads。
(2)使用pyRAD对提取出来的序列进行个体内和个体间的聚类。由于我们关注的是微卫星中重复碱基数目的变异,这一变异类型在聚类时会包含大量的插入/缺失碱基(Indel base)。因此,为了在聚类中尽可能发现所有的微卫星多态性,将分析的聚类阈值(Clustering threshold)、样本内和样本间允许的最大插入缺失(Indel)碱基数分别设置为0.7,40和40。
(3)过滤掉序列中个体间存在碱基转换/颠换的位点。样本间聚类以后,除了微卫星碱基重复数具有差异以外,侧翼序列还存在碱基转换/颠换的位点。若在存在变异位点的侧翼序列区设计引物会影响后续PCR扩增的成功率。因此,使用python脚本将存在碱基转换/颠换的位点过滤掉。同时,设计微卫星引物时需要引物两侧都要具有一定长度的侧翼序列,故只把微卫星重复碱基位于序列中26~129 bp的序列提取出来。这样,微卫星序列的左侧侧翼序列至少有25 bp,右侧侧翼序列至少有21 bp可用于进行后期的引物设计,得出分析的流程图见图1。
(4)使用primer premier 5.0设计引物(Premier Biosoft International,CA,USA)。在设计引物时,只挑选个体间微卫星重复数存在差异的样本进行设计(见图2)。

图1 基于RAD-seq开发SSR引物流程图 reads中红色表示微卫星重复序列Fig.1 Workflow of SSR Primer development based on the reads obtained from RAD-seq Red segment represents the microsatellite sequence in reads

图2 微卫星引物RAD195设计区域Fig.2 Primer region for microsatellite RAD195
1.3 实验验证
为了减少荧光引物合成的成本,在正向引物的5′端添加一个接头序列M13(序列为:5′-TGTAAAACGACGGCCAGT-3′),先用接带M13接头的引物进行扩增,然后添加带荧光的M13序列进行二次扩增[19],PCR扩增体系如下:第一轮PCR扩增体系为:10×PCR Buffer(Mg2+free) 2 μL,dNTP(2.5 mmol·L-1) 0.5 μL,Mg2+(25 mmol·L-1) 2 μL,正向引物(10 μmol·L-1) 0.2 μL,反向引物(10 μmol·L-1) 0.7 μL,Taq酶(5 U·μL-1) 0.2 μL,基因组DNA模板(10 ng·μL-1) 2 μL,牛血清蛋白BSA(20 mg·mL-1) 0.2 μL,ddH2O 12.2 μL,PCR总体积共20 μL。PCR反应程序为:94℃预变性3 min、94℃变性30 s、55℃退火30 s、72℃延伸30 s,35个循环,72℃延伸7 min,12℃保存。第2轮扩增体系为:10×PCR Buffer(Mg2+free) 0.2 μL,Taq酶(5 U·μL-1)0.08 μL,荧光标记M13引物(10 ng·μL-1)0.4 μL,ddH2O 1.32 μL,PCR总体积共2 μL。PCR反应程序为:94℃预变性5 min、94℃变性30 s、53℃退火30 s、72℃延伸30 s,16个循环,72℃最后延伸10 min,12℃保存。
共对3个群体的36个样本进行扩增。扩增结果电泳检测后送美吉生物科技有限公司进行毛细管电泳分型。使用GeneMarker v2.2.0(SoftGenetics,PA USA)对SSR分型结果进行读取和校对。
2 结果与分析
2.1 RAD-Seq测序数据质量评估
所选4个样本的原始序列(Raw reads)数在240~640万条。除去低质量的数据后序列数在180~520万条(见表1)。这些序列中包含微卫星重复片段的序列在46 000~56 000条。
2.2 多态性SSR引物的开发
对样本内进行聚类、过滤掉旁系同源序列后,每个样本包含的位点数在5 546~15 844。样本间聚类后,得到所有样本都具有的位点(一致性位点)1 158个,并得出详细的样本序列信息(见表2)。使用python脚本过滤掉1 158个一致性位点中包含转换/颠换的序列以后,共得到308个位点。进一步过滤掉微卫星重复序列位于reads前25或129 bp以后位置的位点后,得到位点数186个。在这186对引物中挑选出微卫星是2碱基或3碱基重复并最大重复数大于6次,而且在4个样本中至少存在2种不同重复数的引物。共挑选出了40对,使用Primer Premier 5.0设计出了25对引物用于实验验证(见表2)。
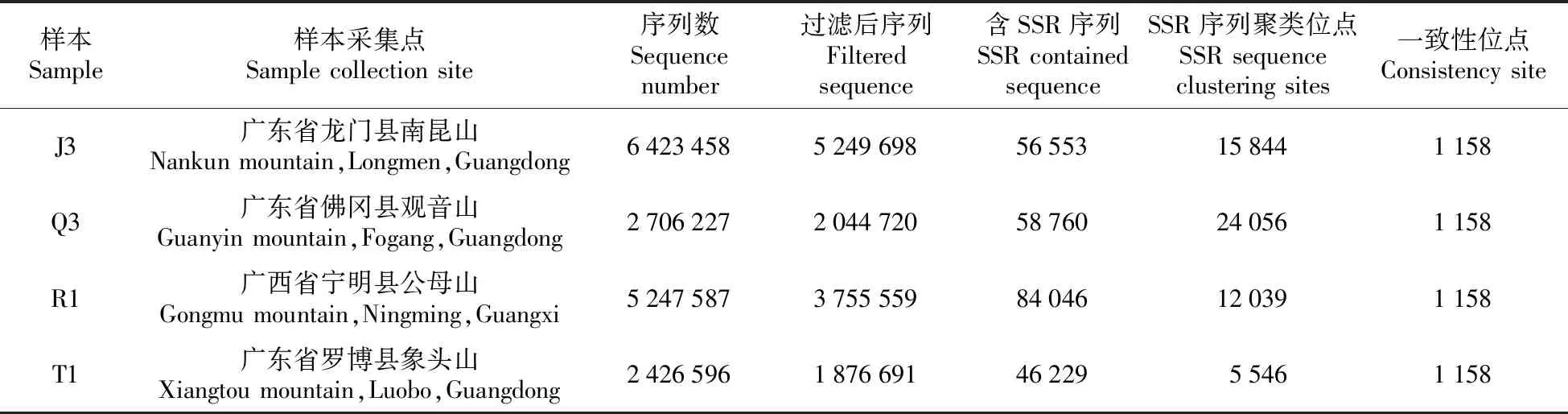
表1 用于SSR引物开发的样本信息
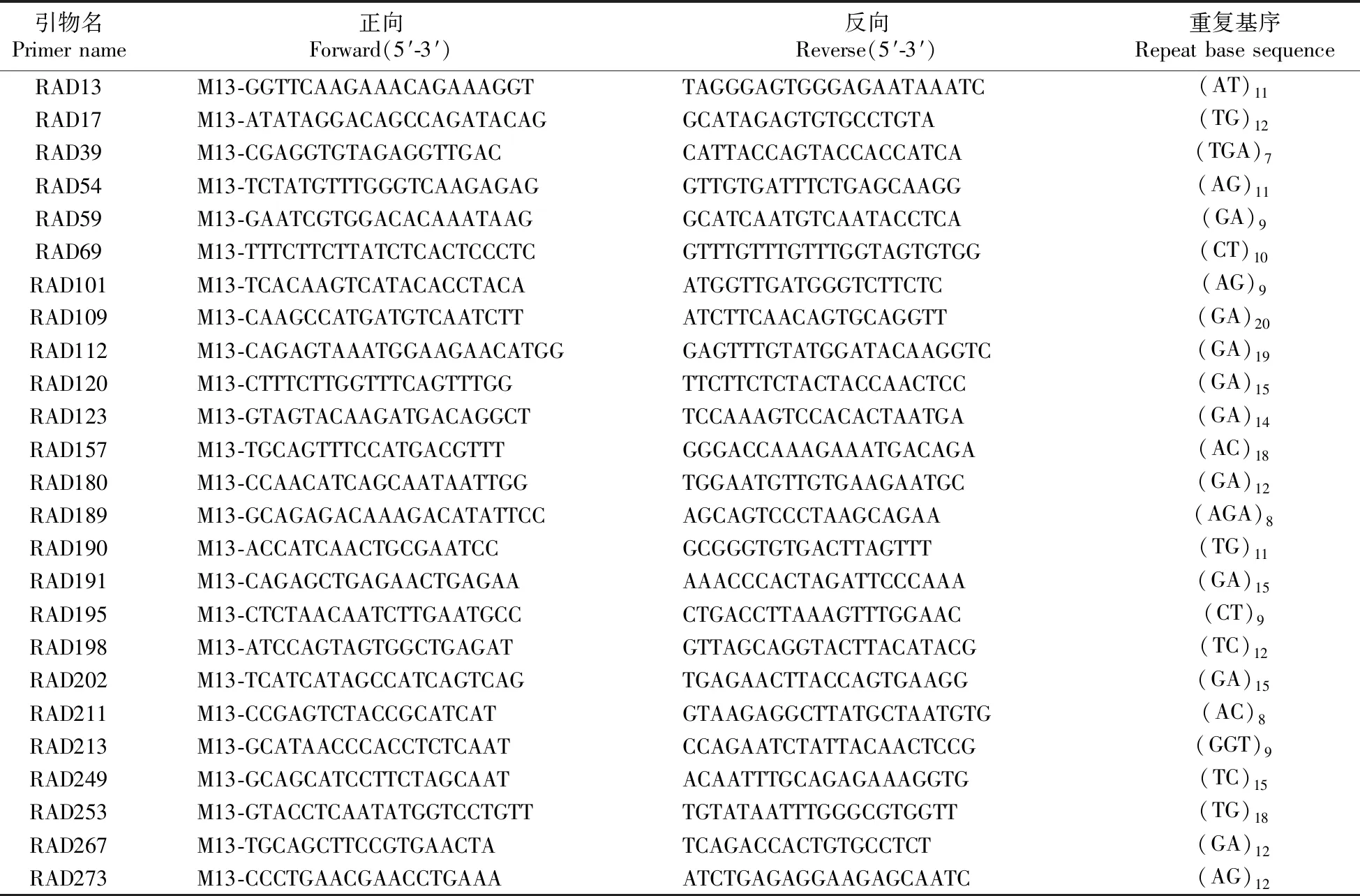
表2 开发用于微卫星多态性筛选的引物序列信息
使用设计好的25对引物对36个个体进行扩增。所有引物的PCR扩增条件和体系见1.3。其中,17对引物被成功扩增出来。这17对引物的等位基因数共为106个,每个引物的等位基因数在2~12,平均为6.2。引物的期望杂合度和观测杂合度分别为0.19~0.88和0.11~0.76。得出引物的详细信息(见表3)。
表3 研究成功扩增引物的多态性信息
Table 3 Polymorphism information of the amplified primers
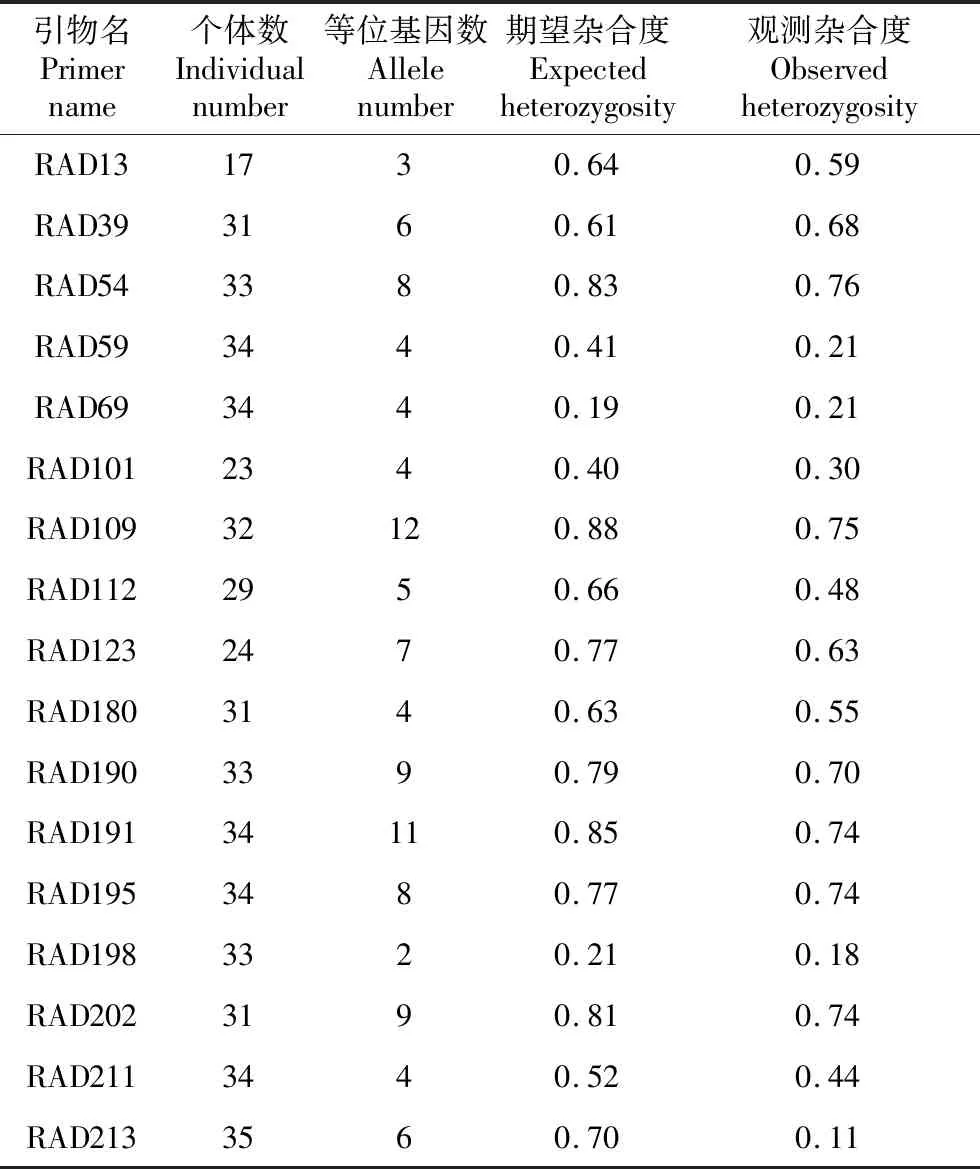
引物名Primername个体数Individualnumber等位基因数Allelenumber期望杂合度Expectedheterozygosity观测杂合度ObservedheterozygosityRAD131730.640.59RAD393160.610.68RAD543380.830.76RAD593440.410.21RAD693440.190.21RAD1012340.400.30RAD10932120.880.75RAD1122950.660.48RAD1232470.770.63RAD1803140.630.55RAD1903390.790.70RAD19134110.850.74RAD1953480.770.74RAD1983320.210.18RAD2023190.810.74RAD2113440.520.44RAD2133560.700.11
3 讨论
本研究基于简化基因组测序方法(RAD-seq)获得的R1端序列成功开发了10多对具有遗传多样性的岭南青冈微卫星引物。根据合成的25对SSR引物,一次性扩增出来17对具有多态性的引物。这17对引物的等位基因数共106个,SSR位点的等位基因数在2~12,平均数为6.2。引物的期望杂合度和观测杂合度分别为0.19~0.88和0.11~0.76。虽然获得的微卫星引物比使用转录组开发的少,但是研究获得的等位基因数目能满足进行物种的群体遗传学、保护生物学及谱系地理学研究的需要。随着测序技术的发展及费用的减低,可以使用PE250对简化基因组数据进行测序。跟本研究采用的PE150测序模式相比,更长读长的PE250测序模式能有更多位点符合微卫星引物的设计。不过对于超大基因组(大于3G)的物种,例如百合科植物的基因组通常达到几十G[20],为了满足测序位点覆盖度的要求可能需要测十几甚至几十G的数据,这极大地增加了测序的成本。因此,我们不建议使用简化基因组测序的方法开发具有超大基因组物种的SSR引物。本次研究中设计的引物都是物种的直系同源序列,但还存在8对引物不能一次性扩增出来。由于正向引物都加了M13接头序列,会对引物的结构和结合特异性有一定影响,可能导致扩增的失败,虽然通过直接合成荧光引物可以消除这一因素的影响,但会增加引物合成的成本。
跟磁珠吸附法开发微卫星引物相比,本次SSR开发方法简化了实验的流程、减少了工作量,并且所需费用也更低。跟利用转录组序列开发微卫星引物相比,使用简化基因组数据的R1端序列开发引物所需要的测序成本、数据的分析能力及计算机资源要求更低[21]。转录组数据的拼接需要计算机具有较大的内存,通常需要服务器才能满足数据分析的需求。本研究的开发方法只需提取含有微卫星位点的序列进行比对即可,这些位点只占所有序列的1%左右。因此,普通的个人电脑即可快速地(1 d以内)完成整个分析。同时,转录组序列位于基因的编码区,比较保守。为了获得足够的遗传变异信息需要进行大量的实验筛选。而简化基因组序列是均匀分布于物种基因组中,具有更高的遗传变异。再次,本研究使用了4个样本进行样本间的聚类,能有效提高筛选出具有多态性的微卫星引物的概率,极大地减少了微卫星引物后期筛选实验的工作量。对于开发引物用于群体遗传学、保护生物学或谱系地理学分析时,此方法可作为有效的候选方法之一。
