余甘子总酚提取纯化工艺的研究
2020-07-08赵春草符晓晖杨清云夏增华张继全
赵春草 符晓晖 杨清云 夏增华 吴 飞* 张继全*
(1.上海中医药大学创新中药研究院, 中药现代制剂技术教育部工程研究中心, 上海201203;2.上海同田生物技术股份有限公司, 上海201203; 3.苏州凯祥生物科技有限公司, 江苏 苏州215600)
藏药余甘子为大戟科植物余甘子Phyllanthus emblicaL.的干燥成熟果实,民间及临床上常将其用于治疗高尿酸血症和痛风,安全性高,不良反应少[1],疗效确切[2-6],主要含有多酚、生物碱、维生素、氨基酸、微量元素[7],性凉,味甘、酸、涩,功效清热凉血、消食健胃、生津止咳。近年来,对余甘子药理活性成分的研究逐渐增多[8-10],发现没食子酸、葡糖倍苷是其主要成分,课题组前期对两者组成的余甘子总酚进行了抗痛风活性研究,确定其具有较好的成药性。
本实验参考中药创新药物研究开发的相关要求,以余甘子多酚成分没食子酸、葡糖倍苷含有量为指标,研究余甘子总酚提取纯化工艺,以期保证原料中间体质量的均一、稳定、可控,为该成分进一步开发成中药有效部位制剂的创新药物奠定基础。
1 材料
1.1 仪器 Agilent 1290型高效液相色谱仪,配置DAD 检测器(美国安捷伦科技公司);XP205型电子分析天平[0.01 mg,梅特勒-托利多仪器(上海) 有限公司];HWS26型恒温水浴锅(巩义市予华仪器有限责任公司);DZF-6050型真空干燥箱(上海精宏实验设备有限公司);SC-150型不锈钢层析柱(江苏沙家浜化工设备有限公司);LD-Y1000A型粉碎机(上海顶帅电器有限公司);R-210型旋转蒸发仪(瑞士Buchi 公司)。
1.2 试剂与药物 余甘子药材(批号150815) 购自安国市一方中药材有限公司,经上海中医药大学中药学院生药教研室倪梁红副教授鉴定为正品。没食子酸对照品(批号110831-201204) 购自中国食品药品检定研究院;葡糖倍苷对照品(批号0761-20140922) 购自上海纯优生物科技有限公司。大孔吸附树脂[型号AB-8,购自天津正天成澄清技术有限公司;型号D101,购自天津农药股份有限公司树脂分公司;型号XAD1600,美国罗门哈斯(陶氏) 公司生产,购自北京慧德易科技有限责任公司;型号Diaion HP-20,日本三菱公司生产,购自北京绿百草科技发展有限公司;型号HZ-801,购自上海华震科技有限公司]。乙腈、甲醇为色谱纯,购自德国Merck 公司;其余试剂均为分析纯,购自国药集团化学试剂有限公司。
2 方法与结果
2.1 总酚含有量测定
2.1.1 对照品溶液制备 精密称取没食子酸对照品适量,50%甲醇定容至10 mL 棕色量瓶中,制得质量浓度1.187 mg/mL 的溶液,精密量取1 mL,50%甲醇稀释至25 mL,即得质量浓度47.48 μg/mL的溶液,同法依次稀释成5.94、23.74、47.48、118.70、237.40、474.80 μg/mL。
精密称取葡糖倍苷对照品适量,50%甲醇定容至10 mL 棕色量瓶中,制得质量浓度1.138 mg/mL的溶液,精密量取1 mL,50%甲醇稀释至10 mL,即得质量浓度113.8 μg/mL 的溶液,同法依次稀释 成 11.38、45.52、113.80、227.60、455.20、910.40 μg/mL。
2.1.2 供试品溶液 精密称取余甘子饮片粉末(过三号筛) 0.15 g,置于具塞锥形瓶中,精密加入50%甲醇50 mL,称定质量,加热回流1 h,放冷,50%甲醇补足减失的质量,微孔滤膜过滤,取续滤液,即得。
2.1.3 色谱条件 ZORBAX Eclipse XDB-C18色谱柱(4.6 mm × 150 mm,5 μm);流动相甲醇(A) -2%乙酸(B),梯度洗脱(0~3 min,95%A;3~10 min,95%~50% A;10~11 min,50%~10%A);体积流量0.8 mL/min;柱温35℃;检测波长273 nm,进样量5 μL。色谱图见图1。
2.1.4 方法学考察
2.1.4.1 线性关系考察 取“2.1.1” 项下2 种对照品溶液,在“2.1.3” 项色谱条件下各进样5 μL测定。以峰面积为纵坐标(Y),溶液质量浓度为横坐标(X) 进行回归,得方程分别为没食子酸Y=19.003X-32.543(r=1)、葡糖倍苷Y=9.685X-36.149(r=0.999 9),分别在5.94~474.80、11.38~910.4 μg/mL 范围内呈良好的线性关系。

图1 各成分HPLC 色谱图Fig.1 HPLC chromatograms of various constituents
2.1.4.2 精密度试验 取1.187 mg/mL 没食子酸对照品溶液、1.138 mg/mL 葡糖倍苷对照品溶液,在“2.1.3” 项色谱条件下进样测定6 次,每次5 μL,测得没食子酸、葡糖倍苷峰面积RSD 分别为0.65%、0.22%,表明仪器精密度良好。
2.1.4.3 重复性试验 取余甘子饮片粉末(过三号筛),按“2.1.2” 项下方法平行制备6 份供试品溶液,在“2.1.3” 项色谱条件下进样测定,测得没食子酸、葡糖倍苷含有量RSD 分别为1.94%、1.00%,表明该方法重复性良好。
2.1.4.4 稳定性试验 取余甘子饮片粉末(过三号筛),按“2.1.2” 项下方法平行制备2 份供试品溶液,于0、2、4、6、8、10、12 h 在“2.1.3”项色谱条件下进样测定,测得没食子酸、葡糖倍苷峰面积RSD 分别为0.21%、0.10%,表明室温下溶液在12 h 内稳定性良好。
2.1.4.5 加样回收率试验 精密称取14 mg 余甘子饮片粉末9 份,精密加入一定量没食子酸、葡糖倍苷对照品溶液,按“2.1.2” 项下方法制备供试品溶液,在“2.1.3” 项色谱条件下进样测定,计算回收率。结果,没食子酸、葡糖倍苷平均加样回收率分别为100.37%、99.80%,RSD 分别为1.24%、0.90%。
2.2 提取工艺考察
2.2.1 单因素试验 取余甘子饮片50 g,以溶剂种类、提取次数、提取时间为影响因素进行单因素试验,每个条件设3组平行对照。虽然水提取、70%乙醇提取时葡糖倍苷、没食子酸转移率均达到最大值,而且基本相同(图2),但水提取时粗提取目标部位纯度较低,故采用乙醇作为提取溶剂。

图2 没食子酸、葡糖倍苷转移率Fig.2 Transfer rates of gallic acid and glucogallin
2.2.2 正交试验 以溶剂用量(A)、提取时间(B)、提取次数(C) 为影响因素,每个因素3个水平;以2 种成分转移率为评价指标,计算综合评分(Y),公式为Y=(没食子酸转移率+葡糖倍苷转移率) ×0.5/100,应用L9(34) 正交试验进行优化。因素水平见表1,结果见表2,方差分析见表3。
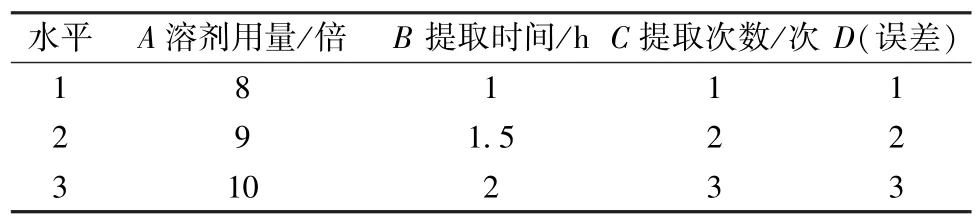
表1 因素水平Tab.1 Factors and levels
由此可知,各因素影响程度依次为提取次数(C) >提取时间(B) >溶剂用量(A),其中C有显著影响(P<0.05),最优提取工艺为A1B3C3,即溶剂用量8 倍,提取时间2 h,提取次数3 次。但在预实验中发现,第3 次提取后引入的杂质量较大,给后续分离纯化工作带来困难,并导致纯化效率降低,故将提取次数暂定为2 次。
2.2.3 提取工艺验证试验 按“2.2.2” 项下优化工艺进行3 批验证试验,每批药材取样量为1 kg,结果见表4。另外还发现第2 次提取时间为1 h 时,提取效率与提取时间为2 h 时差异不大(相对误差约为1%)。最终确定,最优提取工艺为8 倍量70%乙醇回流提取2 次,第1 次2 h,第2次1 h。
2.3 纯化工艺考察 余甘子提取后,粗提物中葡糖倍苷、没食子酸总含有量为6.8%,为达到有效部位含有量不低于50%的要求,需作进一步分离纯化。由于对大孔吸附树脂的研究相对较为成熟,被广泛应用于天然产物的分离和富集[11-14],故本实验应用该方法进行考察。按“2.2.3” 项下优化工艺制备余甘子提取液,浓缩至浸膏,60℃下真空烘干,得到浅褐色干燥固体粉末,收率为48%,备用。

表2 试验设计与结果Tab.2 Design and results of tests

表3 方差分析Tab.3 Analysis of variance
表4 提取工艺验证试验结果(n=3)Tab.4 Results of verification tests for extraction process(n=3)
2.3.1 大孔吸附树脂
2.3.1.1 种类 具有大孔结构的非极性大孔吸附树脂可吸附极性较小的有机化合物[15],本实验选择常用型号(AB-8、D101、XAD1600、HP-20、HZ-801),检测其吸附量、解吸率,结果见表5。最终,选择D101 大孔吸附树脂作进一步分离纯化工艺研究[16-17]。

表5 大孔吸附树脂吸附量、解吸率测定结果Tab.5 Results of adsorption quantity and desorption rate determination of macroporous adsorption resins
2.3.1.2 层析柱径高比 称取预处理后的D101树脂400 g,平均分为4 份,每份100 g,湿法装柱。取4 份“2.3” 项下药材粉末,加适量水制成0.2 g/mL 溶液,以1 BV/h 体积流量上样,考察径高比1∶3、1∶6、1∶10、1∶15 对没食子酸、葡糖倍苷吸附率的影响,结果见图3。由此可知,柱径高比为1∶6 时2 种成分吸附率最高,故选择其作进一步研究。
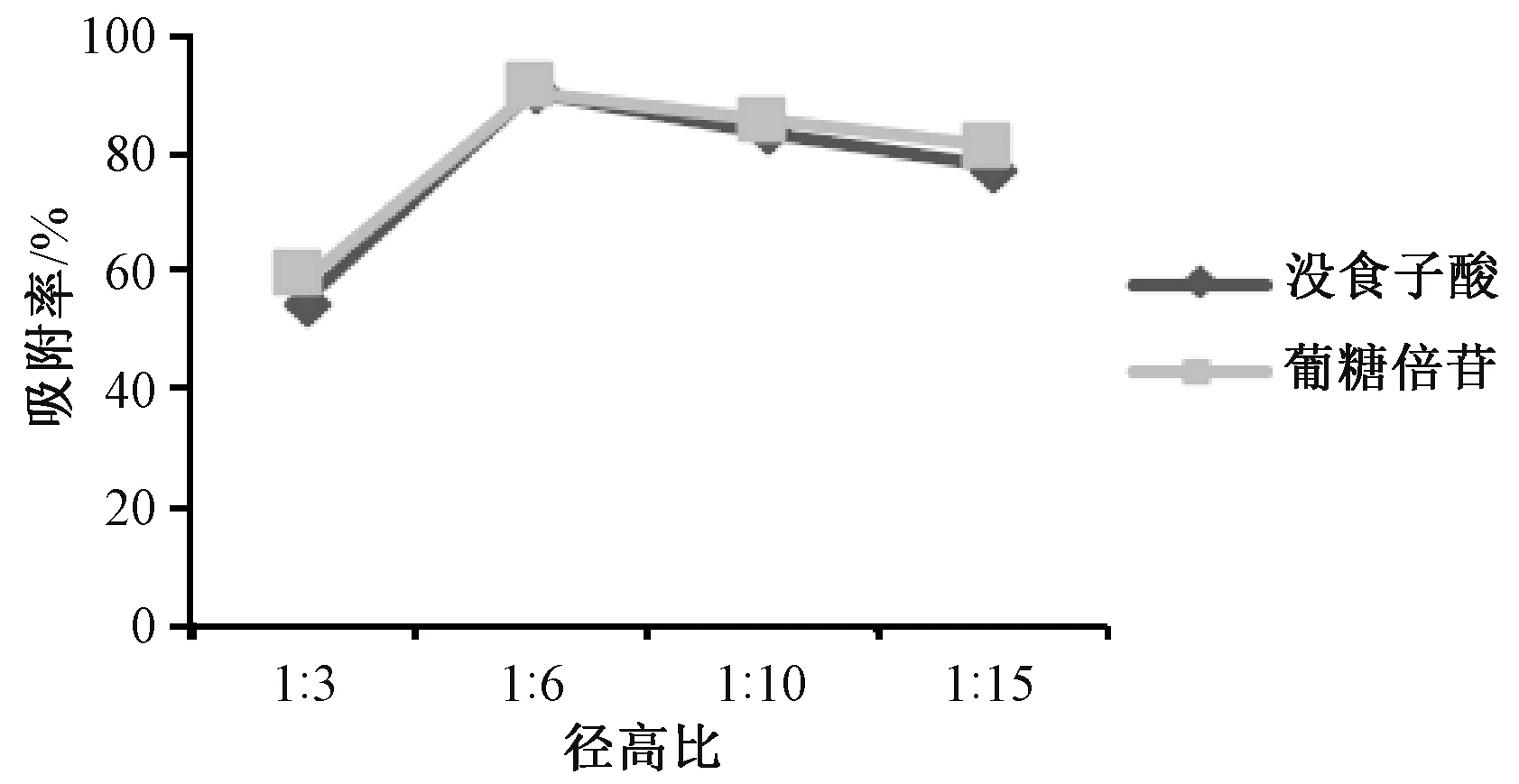
图3 径高比对没食子酸、葡糖倍苷吸附率的影响Fig.3 Effects of diameter-height ratio on the adsorption rates of gallic acid and glucogallin
2.3.2 上样药液
2.3.2.1 上样质量浓度 称取预处理后的D101树脂100 g,湿法装柱,取“2.3” 项下药材粉末,加适量水制成一定质量浓度溶液,以1 BV/h 体积流量上样,考察上样质量浓度0.05、0.1、0.2、0.3、0.4 g/mL 对没食子酸、葡糖倍苷吸附率的影响,结果见图4。由此可知,上样质量浓度为0.2 g/mL时2 种成分吸附率最高,故选择其作进一步研究。

图4 上样质量浓度对没食子酸、葡糖倍苷吸附率的影响Fig.4 Effects of sample concentration on the adsorption rates of gallic acid and glucogallin
2.3.2.2 上样体积流量 取“2.3” 项下药材粉末,加适量水制成0.2 g/mL 溶液,考察体积流量0.5、1、1.5、2、2.5 BV/h 对没食子酸、葡糖倍苷吸附率的影响,结果见图5。由此可知,上样体积流量越小吸附越充分,但周期也随之延长,综合考虑时间、吸附率,选择1 BV/h作进一步研究。

图5 上样体积流量对没食子酸、葡糖倍苷吸附率的影响Fig.5 Effects of sample volumetric flow rate on the adsorption rates of gallic acid and glucogallin
2.3.3 洗脱工艺 本实验研究目的是确定合适的洗脱速率梯度,使水洗脱过程中水溶性杂质与目标部位达到良好的分离,并且目标部位活性组分含有量符合相关要求。
2.3.3.1 洗脱体积流量 称取预处理后的D101树脂100 g,平行5 份,湿法装柱,径高比为1∶6。取上样液80 mL(没食子酸、葡糖倍苷总含有量为10.64%),400 mL 纯化水以不同体积流量进行洗脱,前1.5 BV 洗脱液弃去,收集第1.5~4 BV 洗脱液,浓缩干燥,测定2 种成分总含有量,结果见图6。由此可知,洗脱体积流量越大,总含有量越低,但过大时水溶性杂质与目标组分不能得到很好的分离,从而影响终产品目标组分含有量,最终确定为1.5 BV/h。
2.3.3.2 加水量 称取预处理后的D101 树脂500 g,湿法装入层析柱中,取“2.3” 项下药材粉末,配制成0.2 g/mL 药液100 mL,以1 BV/h 体积流量通过层析柱,用水洗脱,每半个柱体积收集1 份,直至无目标组分流出,再用50%、95%乙醇进行洗脱,接收液分别浓缩、干燥后得到固体粉末,测定没食子酸、葡糖倍苷总含有量,结果见图7。由此可知,当加水量达4 BV 时,目标组分已基本上完全被洗脱。合并1.5~4 BV 洗脱液,浓缩干燥后测得2 种成分总含有量为57.9%,符合预定要求,故选择4 BV 水洗脱比较合适(前1.5 倍柱体积所含目标成分纯度不高,弃去)。
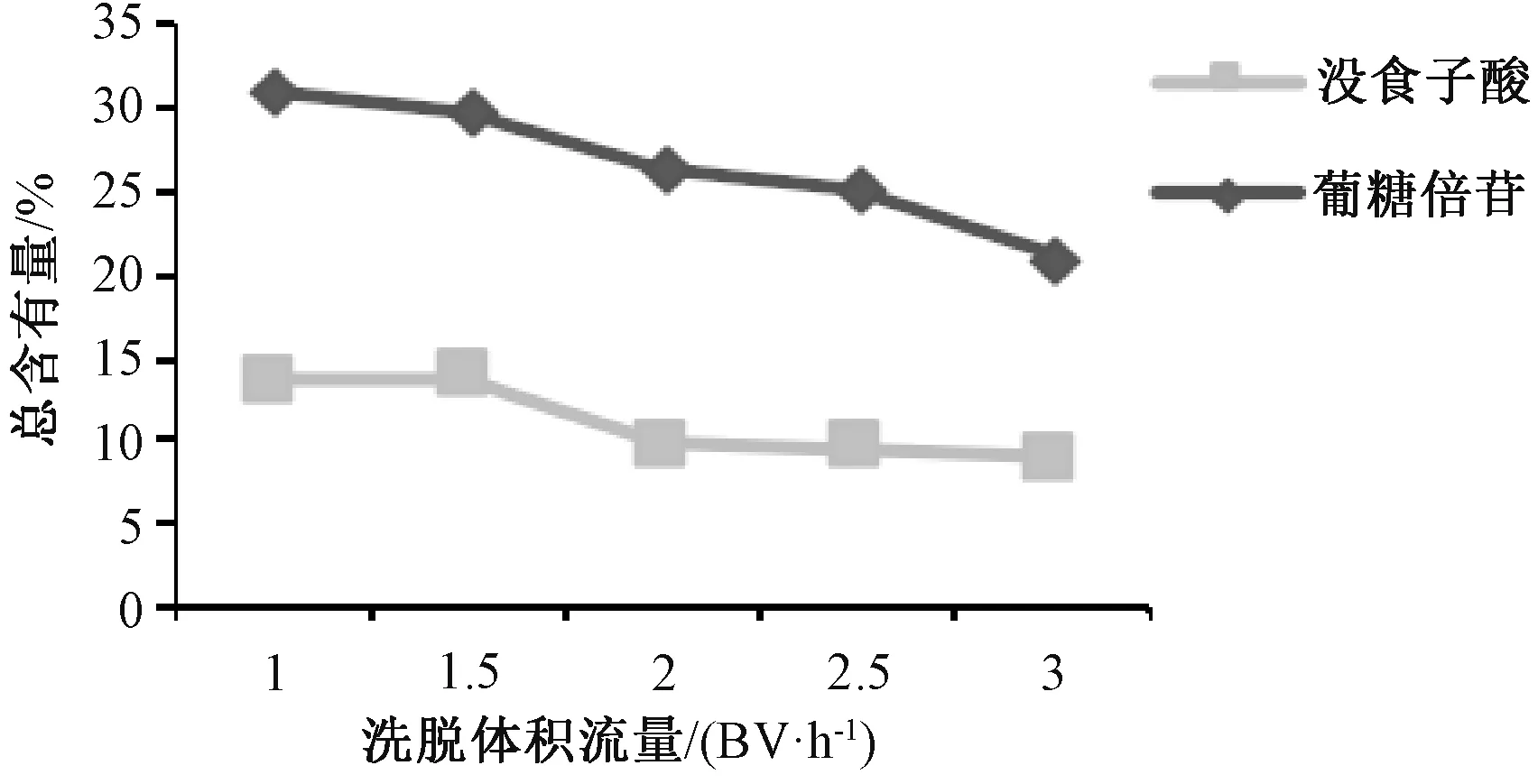
图6 洗脱体积流量对没食子酸、葡糖倍苷总含有量的影响Fig.6 Effect of elution volumetric flow rate on the total content of gallic acid and glucogallin

图7 加水量对没食子酸、葡糖倍苷总含有量的影响Fig.7 Effect of water consumption on the total content of gallic acid and glucogallin
2.3.4 分离纯化工艺确定 选用经预处理的D101 树脂,柱径高比为1∶6,上样质量浓度为0.2 g/mL,上样体积流量为1.5 BV/h,提取物用水溶解上柱,每500 g 树脂先用750 mL(1.5 BV) 水洗脱除去部分非目标杂质,再用1 250 mL(1.5~4 BV) 水洗脱收集,浓缩干燥,得到余甘子总酚,其性状为白色至淡黄色固体粉末,收率约为4.8%。
2.4 纯化工艺验证试验 通过3 批验证试验可知,余甘子总酚有效部位中没食子酸、葡糖倍苷总含有量均在50%以上,见表6。
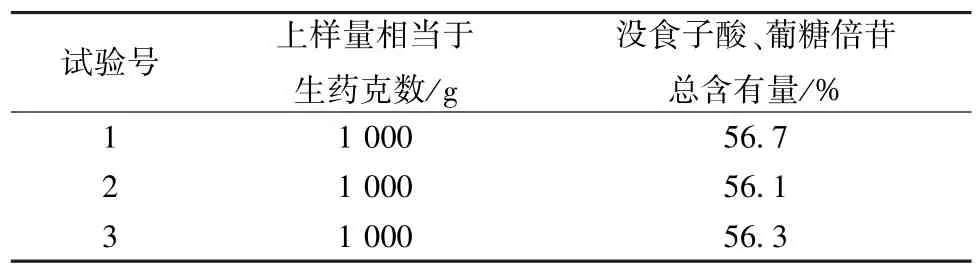
表6 纯化工艺验证试验结果(n=3)Tab.6 Results of verification tests for purification process(n=3)
3 讨论与结论
根据《药品注册管理办法》 的相关要求,中药有效部位制剂定义为“未在国内上市销售的从植物、动物、矿物等物质中提取的有效部位及其制剂,其有效部位含量应占提取物的50%以上”。为了提高有效部位原料的质量可控性、安全性、有效性,本研究以HPLC 法测定已知结构化合物含有量大于50%为目标,发现相比于紫外色谱法,有效部位含有量的要求进一步提高。
综上所述,本实验通过筛选大孔吸附树脂材料、优化余甘子总酚提取纯化工艺,结合多批中试验证,确定了抗痛风中药有效部位制备工艺路线及参数,并参考了中药有效部位新药注册申请的相关要求。上述研究过程和思路具有一定的代表性,可为同类新药制备工艺的开发提供参考和借鉴。
