Therapeutic effect of hydrogen peroxide via altered expression of glutathione S-transferase and peroxiredoxin-2 in hepatocellular carcinoma
2020-07-07ZehraHashimAmberIlyasShamshadZarina
Zehra Hashim , Amber Ilyas , Shamshad Zarina
Dr. Zafar H. Zaidi Center for Proteomics, University of Karachi, Karachi 75270, Pakistan
Keywords:Oxidative stress Hydrogen peroxide GSTP1 Prx2 Hepatocellular carcinoma
A B S T R A C T Background: Hepatocellular carcinoma (HCC) has a high incidence and mortality that epitomizes one of the prominent causes of cancer-related death globally. Novel therapeutic approaches are therefore required. Reactive oxygen species (ROS) are necessary for maintaining cell cycle. Although ROS is involved in HCC progression, hydrogen peroxide (H 2 O 2 ) has anti-proliferative effect on HCC.Method: HCC Huh-7 cells were cultured and incubated with various concentrations of H 2 O 2 . Paraoxonase activity, levels of malondialdehyde, glutathione and protein oxidation were measured in treated and untreated Huh-7 cells. Furthermore, untreated and treated Huh-7 cells were subjected to two dimensional gel electrophoresis and identified protein spots which were differentially expressed by LC-MS/MS analysis. qRT-PCR was performed to validate the identified proteins.Resul ts: depleted glutathione (GSH) with the concomitant up-regulation of GSTP1 and Prx2. also increased malondialdehyde and protein oxidation, decreased the activity of paraoxonase in Huh-7 cells.Conclusion: H 2 O 2 could be used as a novel therapeutic agent that might be beneficial in inducing cell cytotoxicity and hence suppress HCC proliferation.
Introduction
Oxygen is essential for life yet also inherently toxic for life,and this phenomenon is referred as oxygen paradox [1] . During normal metabolism, more than 95% of oxygen free radicals such asetc. are produced by electron transport chain [2] .Free radicals are important for maintaining cell cycle by influencing several cellular processes including signal transduction,defense against foreign invaders like bacteria, viruses, etc. [3] .Under normal circumstances, our body is capable of dealing with production of reactive oxygen species (ROS). Excessive production of ROS may result in potentially cytotoxic effect called oxidative stress [4] .
At pro-oxidant state, free radicals cause damage to proteins,lipids, RNA and DNA components, which may cause cellular lesion [5] . It has been well established that oxidative stress may contribute to the progression of many diseases including cardiovascular disease, hypertension, atherosclerosis, diabetes, and cancer [6] .Oxidative stress is suggested to be a causative factor in the progression of cancer [7] hence emphasizing importance of antioxidant system that may prevent oxidative damage and cancer [8] . It has been observed that oxidative stress has a significant contribution to the development and advancement of hepatocellular carcinoma (HCC) [9] .
Recent studies suggest that pro-oxidant therapies can induce mitochondrial depolarization and exert cytotoxic effect on cancer cells [10 , 11] . Previously it has been reported that increased amount of ROS cause necrosis and at lower concentrations instigate apoptosis [12] . Pro-oxidant agents increase intracellular ROS that initiate oxidative metabolism producing cytotoxic oxidative stress in cancer cells resulting in collapse of defense mechanism, causing DNA damage and hence apoptosis [13-15] .
Besides DNA damage, ROS also cause impairment of cellular protein and membrane lipids [16] . Free radicals induce lipid peroxidation via reaction with polyunsaturated fatty acids. Elevated levels of ROS inhibit tumor cell growth and enhance apoptosis in cancerous cells by lipid peroxidation [17] .
Oxidative stress has been measured by oxidative stress biomarkers including antioxidant enzymes [18] such as glutathione peroxidase, superoxide dismutase, catalase, paraoxonase, etc. and lipid peroxidation product such as malondialdehyde (MDA) that is hydrolyzed by paraoxonase [19 , 20] . It has been reported that paraoxonase activity is inversely correlated with ROS-induced lipid oxidation. Paraoxonase has anti-oxidative stress effect [21] .
Glutathione S-transferases (GSTs) play a crucial role in detoxification against oxidative damage [22] . Among all GSTs, Glutathione S-Transferase Pi 1 (GSTP1) is involved in cellular existence and apoptotic cell death via mitogen-activated protein (MAP) kinase pathway. GSTP1 is an inhibitor of c-Jun N-terminal kinase 1 (JNK1)which in turn regulates cell cycle against stress response [23] . It has been demonstrated that excessive ROS production up-regulates GSTP via the JNK pathway that leads to cell death [23] .
Peroxiredoxin family consists of six members that regulate antioxidant defense, drug resistance and apoptotic cell death in various cancers [24] . It has been observed that different peroxiredoxin is downregulated in malignant cells [25 , 26] . Ralat et al. [27] revealed that oxidative stress activates peroxiredoxin which is associated with GSTP1 elevation that consequently acts on hydroperoxides.
Apoptosis or c ell death in cancer cells can be induced by various chemical compounds such as oxidized LDL, dithiothreitol (DTT)and[28-30].effects on cancer cells in different ways including inflammation, cell cycle arrest, DNA damage, migration and apoptosis [31]. It has been suggested thatexhibits antiproliferative activity and can cause oxidative stress which mediated cell death in cancer [32-34], hence can be beneficial as therapeutic agent [35]. In mitochondria,can induce disrupted cellular signaling that leads to cell cycle arrest and hence senescence. Due to aberrant cell signalingmight suppress tumor growth [36] .Thereforecould be a promising candidate that increases ROS levels and can damage cancer cells.
In current study, we have experimented the effect ofon HCC cells to examine the involvement of oxygen free radicals in cancer cell death. Furthermore, we have identified potential oxidative stress biomarkers intreated and untreated HCC cells as potential therapeutic target to observe the response of oxidative stress in reducing growth of cancer cells.
Methods
CellcultureandincubationwithH2O2
Human HCC Huh-7 cell line were cultured in Duibecco’s Modified Eagle’s Medium supplemented with 2 mmol/L glutamine,10% fetal bovine serum, 4.5 g/L glucose, 100 U/mL penicillin and 100 μg/mL streptomycine. Cells were grown in a incubator (Hera-Cell 150, Thermo Heraeus) with 5% CO 2 at 37 °C. In exponential growth phase cells were incubated with different concentrations of H 2 O 2 (10, 20, 30, 40, 50, 100 and 150 μmol/L).treatments were conducted for 60 min in triplicates.
Cytotoxicityassay
HCC Huh-7 cells (1.2 ×106cells/mL) were plated in 6-well plates (Corning, New York, USA). Cells were allowed to attach for 24 h with 5%at 37 °C before cytotoxicity assay. After 24 h,fresh medium with various concentrations of the(10, 20, 30,40, 50, 100 and 150 μmol/L) were added for 1 h. Followed by addition of fresh medium, cells grew for another 24 h. Media were taken from each treatment and cytotoxicity assay was carried out using Cell Cytotoxicity Assay Kit (Abcam, Massachusetts, USA) as per manufacturer’s protocol. Absorbance at 490 nm was recorded using microplate reader (Backmann Coulter, California, USA) to calculate cytotoxicity. Growth of cells relative to their respective controls was expressed as percent cell death. All assays were run in triplicate.
Paraoxonase
Paraoxonase activity was measured as previously described [37] .The assay mixture containedand paraoxon 1.0 mmol/L each in 50 mmol/L glycine/NaOH buffer pH 10.5. An aliquote of cell lysate was used and the increase in absorbance was recorded at 412 nm that was due to the formation of 4-nitrophenol.Paraoxon was used as a substrate and molar extinction coefficient 18,290 M−1cm−1was used to calculate paraoxonase activity.
Malondialdehyde
Level of MDA was determined by estimating the colored product formed by the reaction between thiobarbituric acid and MDA as described previously [38] . An aliquote of cell lysate was treated with 20% trichloroacetic acid, 8.1% SDS and aqueous 0.8% TBA. The assay mixture was placed on boiling water bath and heated for 60 min then cooled on ice. N-butanol was then added to the assay mixture and centrifuged for 10 min at 40 0 0 rpm. Organic layer containing reaction product was separated and read at 532 nm.Standard curve was prepared using 1,1,3,3-Tetraethoxy propane.
Glutathione
Content of reduced glutathione (GSH) was measured by Ellman’s method [39] . Briefly, 1 mL cell lysate was mixed with 10%trichloroacetic acid and then centrifuged at 50 0 0 rpm. Supernatant was collected and deproteinized using 1 mL of 5,5-dithiobis 2-nitrobenzoic acid (Ellman’s reagent) solution. The yellow colored product was read at 412 nm. GSH was used as standard and amount of GSH was expressed as μmol/μg protein.
Proteinoxidation
Protein carbonyl content was measured as described by Levine et al. [40] . A total of 0.5 mL cell lysate was treated with 2.0 mL of 10 mmol/L 2,4-dinitrophenylhydrazine (DNPH). A control sample was also prepared in which 2.5 mol/L HCl was added. In order to obtain protein pallet, sample was treated with 20% trichloroacetic acid (TCA) followed by centrifugation at 3500 rpm for 20 min.The pallet was again washed with 10% TCA. Final precipitates were washed with ethanol: ethyl acetate (1:1 v/v). After centrifugation protein pallet was resuspended in 6 mol/L guanidine hydrochloride and incubated for 10 min at 37 °C. Samples were read at 370 nm against reagent blank. Absorption coefficient (e)of 22,0 0 0 M-1cm-1was used to calculate protein carbonyl content and was expressed as nmol/mg protein.
Two-dimensionalgelelectrophoresis
Extraction of total protein was performed from cells using lysis buffer that contain DTT 0.5 mol/L, urea 2 mol/L, EDTA 0.2 mol/L,NP40, glycerol, ampholyte (pH 3-10) and protease inhibitor cocktail in 0.5 mol/L Tris-HCl buffer. Protein concentration was measurement by BCA protein assay kit (Pierce). Extracted protein were dissolved in rehydration buffer and applied on 3-10 NL, 7 cm IPG strips for 16 h at room temperature for passive rehydration. Isoelectric focusing (IEF) was performed on IEF Multiphor II system(GE Healthcare) at 20 °C using total of 11,0 0 0 V/h. Followed by IEF,IPG strips were incubated with equilibration buffer I and II containing DTT and iodoacetamide, respectively. After equilibration,IPG strips were subjected for second dimension on 12% SDS-PAGE.Protein spots were visualized on gels after staining with coomassie blue G-250. Two-dimensional gel electrophoresis (2DE) was performed in triplicate.
2D-gelanalysis
To perform the differential protein expression, 2DE gels were analyzed with PDQuest software (Bio-Rad). All the gels from untreated control and treated groups were matched. Intensity of each protein spot was determined by subtracting background intensity. The spot intensity was calculated and presented as mean ± SD. APvalue of<0.05 was considered statistically significant.
ProteinidentificationbyLC-MS/MS
Selected protein spots were excised from the gel using EXQuest spot cutter (BioRad, California, USA) and washed with Acetonitrile and milliQ water (1:1). The gel pieces were become white followed by rehydration with 100 mmol/L ammonium bicarbonate. The gel spots were vacuum dried using Eppendorfvacufuge vacuum concentrator. Dried gel spots were incubated first with 10 mmol/L DTT at 60 °C for 45 min and then with Iodoacetamide 55 mmol/L for 20 min in the dark at room temperature. Followed by reduction and alkylation, the gel spots were digested with 2 ng/μL trypsin at 37 °C overnight. Twenty-five mmol/L ammonium bicarbonate and 10% formic acid were used to extract peptides. Desalting was performed using zip tip and peptides were vacuum dried.
Purified peptides were reconstituted with 0.1% formic acid (FA).Digested peptides were applied on C-18 nano column (75 μm ID ×15 cm Pep-Map 100) at constant flow rate 300 nL/min. 0.1%FA as buffer A and 98% ACN, 2%, 0.1% FA as buffer B were used to equilibrate the column, where A is 96.8% and B is 3.2%.A multi-step gradient for buffer B from 3.2% to 80% was used over 70 min. Tandem MS analysis was conducted using linear ion trap Mass spectrometer (Thermo LTQ XL, Thermo Fisher Scientific, Massachusetts, USA). BSA (66 kDa), myoglobin (17.2 kDa) and cytochrome c (12.3 kDa) were used as standards. Proteome Discoverer 1.2 was used to convert raw files into MGF files. Mascot search engine was used for protein identification against UniProtKB database. Search parameters were as follows. Fixed modification:carbamidomethylation of Cys; variable modification: met oxidation; peptide mass tolerance: ±1.5 Da; MS/MS tolerance: ±0.5 Da;missed Cleavages: 1.
Quantitativereversetranscriptionpolymerasechainreaction(qRT-PCR)
Total RNA was isolated from cells using TRI Reagent(Sigma -Aldrich, Missouri, USA) according to protocol provided by manufacturer. Purity of the extracted RNA was checked by taking the 260/280 OD ratio. RevertAid First-Strand cDNA synthesis kit(ThermoFisher Scientific) was used to reverse transcribed 500 ng RNA into cDNA. Primers that correspondPeroxiredoxin2(Prx2),GlutathioneS-transferasep1(GSTP1)andNuclearfactorerythroid2-relatedfactor2(NFE2L2)genes were used for amplification. Reaction mixture for real-time PCR amplification contains cDNA, SYBR green master mix, primers and DNase/RNase free water. Following conditions were used for qRT-PCR 7300 system (Applied Biosystems Corp., California, USA); 1 cycle for 5 min at 50 °C, 1 cycle for 10 min at 95 °C and 40 cycles for 14 s at 95 °C for denaturation, 1 min at 55 °C for annealing and 45 s at 72 °C for extension.Analysis for dissociation curve was also conducted.Glyceraldehyde-3-phosphatedehydrogenase(GAPDH)gene was used for data normalization. Relative expressions of thePrx2andGSTP1were evaluated with relevance to the expression in untreated Huh-7 cells that utilize ddCt relative quantification method.

Fig. 1. Cytotoxic response of Huh-7 cells incubated with different concentrations of H 2 O 2 . Cellular cytotoxicity was expressed as mean ±SD versus cells in culture medium without H 2 O 2 (0% cytotoxicity). Data have been collected from triplicate experiments. Differences between untreated and H 2 O 2 treated cells were significant for all concentrations of H 2 O 2 . ∗: P < 0.05 versus basal level.
Protein-proteininteraction(PPI)analysis
Interaction of oxidative stress related proteins were determined and correlate with identified Prx2 and GSTP1. Analyses of PPI was predicted using STRING software ( http://string-db.org ).
Statisticalanalysis
SPSS software (SPSS for Windows 20.0) was used for data analyses. Data was presented as mean ±SD. One-way ANOVA was applied for group comparisons. Whenever appropriate Post-hoc test was performed by Bonferoni’s method. All the experiments were run in triplicate. APvalue of<0.05 was considered statistically significant.
Results
effectsinvitro
Treatment of Huh-7 cells with rising concentrations ofdecreased the cell proliferation and increased cytotoxicity in dosedependent manner ( Fig. 1 ). At concentration varying from 10 to 150 μmol/L,increased the cytotoxicity rate up to 30% within 60 min (P<0.05). Cytotoxicity was found to be negligible at 10 μmol/L. Maximum inhibition (30%) was observed at 150 μmol/L concentration. Results of our study proposed 150 μmol/L concentration as the most efficient because at this concentration cytotoxicity markedly increased up to 30% hence it was used for further studies.
Anti-oxidantstatus,MDAandproteinoxidation
Paraoxonase activity, level of GSH, MDA and protein oxidation intreated and untreated control Huh-7 cells are presented in Fig. 2 . Paraoxonase activity and GSH were significantly (P<0.05)lower inincubated cells with a concomitant rise (P<0.05)in protein oxidation and MDA as compared to untreated control Huh-7 cells.

Fig. 3. The corresponding expression of GSTP1 and Prx2 in H 2 O 2 treated and untreated Huh7 cells. ∗represents statistically significant value ( P < 0.05). Each bar represents mean ±SD. All experiments were run in triplicate.
DifferentialexpressionofPrx2andGSTP1inHCCHuh-7cells

Fig. 4. Two-dimensional gel electrophoresis profile of proteins from untreated control ( A ) and 150 μmol/L H 2 O 2 treated Huh-7 cells ( B ). Labeled spots are identified by MS/MS analysis.

Fig. 5. Intensity of Prx2, GSTP1 and NFE2L2 protein spots in H 2 O 2 treated and untreated human HCC Huh-7 cells analyzed by PD Quest software. ∗: P < 0.05 compared with untreated cells.
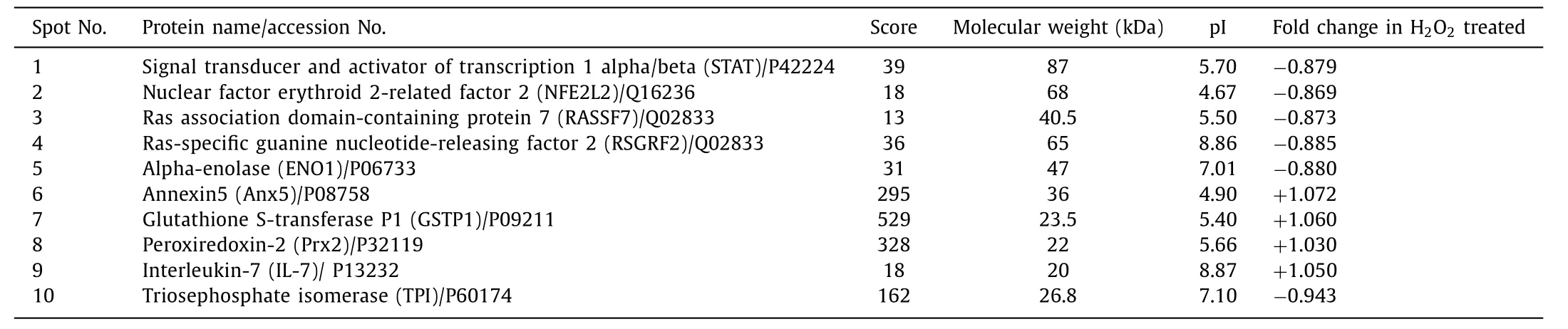
Table 1 Differentially expressed proteins from H 2 O 2 treated and untreated Huh-7 cell line identified with nano LC-MS/MS.
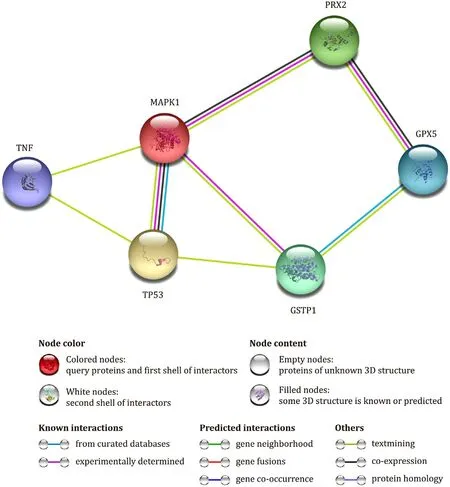
Fig. 6. Protein-Protein Interaction analysis using String software. Interaction between peroxiredoxin-2 (PRDX2), MAP kinase1 (MAPK1), glutathion peroxidase (GPX5), glutathion S transferase P1(GSTP1), tumor protein 53 (TP53) and tumor necrosis factor (TNF) are presented.
Identificationofdifferentiallyexpressedproteins
Two dimensional gel electrophoresis and mass spectrometry were used to determine the differential protein expression intreated Huh-7 cells. 2DE profile of untreated control Huh-7 cells andtreated cells was presented in Fig. 4 A and B, respectively.Protein spots with differential expression were identified through nLC-MS/MS analysis as shown in Table 1 and Fig. 4 . GSTP1, Prx2 and NFE2L2 are of major interest due to their involvement in the management of oxidative stress. The intensities of identified spots intreated Huh-7 cells are shown in Fig. 5 . We have observed up-regulation of GSTP1 and Prx2 proteins, and down-regulation of NFE2L2 protein intreated Huh-7 cells as compared to untreated cells.
Discussion

Fig. 7. Flow diagram indicating oxidative stress induced tumor suppression. Solid arrows indicate the levels predicted in H 2 O 2 induced oxidative stress in Huh-7 cells. GSH:reduced glutathione; Prx: peroxiredoxin; GSTP: glutathione S-transferase protein; MDA: malondialdehyde.
Oxidative stress activates antioxidant defense mechanism to minimize the damage effect. However, higher concentration of ROS may devastate the cells. Elevated level of free radicals that surpasses the protection limit of antioxidant causes oxidative damage and hence cell apoptosis [41 , 42] . Previous studies demonstrated that H 2 O 2 induced oxidative stress causes cell death [43-45] . During various clinical conditions like Alzheimer’s [46] , cardiovascular diseases [47] , rheumatoid arthritis, and cancer, ROS performs an important role in cell cycle regulation [48] . As ROS synthesized in mitochondria which is also a target, it escorts free radicals to damage membrane phospholipids hence mitochondrial membrane potential is lost. Due to reduced membrane potential, mitochondria releases cytochrome c that ultimately triggers the activation of caspase-3. This leads to DNA damage resulting in cell apoptosis [49-52] . In the present study we have found decreased levels of GSH in H 2 O 2 incubated cells as compared to untreated control. Schumacker [53] reported that oxidative stress induced ROS generation lead to the depletion of GSH hence inhibit tumorigenesis. GSH is responsible for redox status of cell [54] and is present in high concentration in cytosol whereas only 10% −15%GSH is found in mitochondria [55] . GSH is made up of cysteine,glycine and glutamic acid and forms a complex called glutamate cysteine ligase (GCL). The GCL comprises of two components GCLC and GCLM catalytic and amplifier, respectively. Primary function of GSH is to support antioxidant enzymes that scavenge H 2 O 2 .Harris et al. [56] used oncogene-induced murine models of mammary cancer that were deficient of GCLM. Lack of GCLM caused 75% reduction in GSH leading to intensified oxidative damage. As a result of persistent oxidative stress, tumor progression is inhibited compared to mice with normal GSH level, suggesting that oxidative stress intervened by GSH reduction may slow down tumor growth. Besides cell protection from deleterious effect of ROS [57] ,GSH also plays a crucial role in regulation of gene transcription and apoptosis [58 , 59] .
Furthermore, we have also observed significantly increased expressions of protein and mRNA levels of GSTP1 and Prx2, and significantly decreased expression of NFE2L2 protein and mRNA intreated HCC Huh-7 cells. Prx family is proposed to regulate cellular redox status. Among all members of Prx family, Prx2 protects cells from oxidative stress [60] . In various diseases including cancer [61] , Prx2 is found to be elevated to respond to the increased ROS levels hence offering a protective role. In contrast to the suggestion that oxidative stress contributes towards cancer progression [62] , we found that increased production of ROS causes oxidative damage that eventually triggers cell cytotoxicity. Similar results have been reported earlier [15 , 41 , 63] . Oxidative stress induced byactivates nuclear factor kappa B (NF-κB) which is a redox sensitive transcription factor entailed in cell propagation,and may stimulate Prx2 level [64] . It has been demonstrated that Prx2 suppresses the growth of smooth muscle cells and suppresses platelet-derived growth factor signaling [65] . It has also been suggested that Prx2 inhibits the cell proliferation in leukemia and acts as a growth suppressor [66] .
In current study we also observed up-regulation of GSTP1 at protein and mRNA levels intreated Huh-7 cells. GSTP1 is a member of GSTs family. Along with other members of GSTs,GSTP1 is involved in the detoxification of free radicals and protect cells from oxidative damage [67] . It has been reported that ininduced oxidative stress, GSTP1 activates Prx2 and their elevated levels act onor hydroperoxides [27] . In the presence of ROS, GSTP1 is up-regulated and conjugates electrophiles with reduced glutathione to minimize oxidative stress [68] . Under normal physiological conditions GSTP1 forms a protein complex with JNK and inhibits it to regulate the apoptosis [69] . During stress, GSTP1 dissociates itself from JNK and becomes inactive [70] .While JNK restores its activity by phosphorylation and hence increases the mRNA ofAP-1genes. As a result, signaling cascades for stress response are activated consequently inducing apoptosis( Fig. 6 ) [71 , 72] . Nuclear factor erythroid 2-related factor 2 (NFE2L2)is another identified protein that plays an important role in oxidative stress regulation. Keap1-NFE2L2-ARE is a crucial signaling pathway for maintaining redox balance. Under oxidative stress, Keap1-NFE2L2-ARE pathway performs adaptive role,in absence of which oxidative stress cause uncontrolled cell proliferation [73-75] . Our results are also in agreement with previous study that demonstrates down-regulation of NF2L2 as a result of increased level of oxidative stress due toinduction [76] .
ROS are responsible for DNA, protein and lipid damage ( Fig. 7 ).In cells proteins are major targets of oxygen free radicals. In compliance with earlier study [77] we also found elevated level of protein carbonyl contents intreated cells. Oxidized proteins accrue during aging and in some clinical disorders [78] . ROS have potential to cleave peptide backbone via oxidizing amino acids such as proline, lysine and arginine. Different mechanisms such as formation ofα-amidation pathway, protein-protein cross-linked derivatives, damage of cell membrane and cleavage of glutamyl residues occurring by lipid oxidation products [79] are involved in oxidative cleavage that provides reactive ketones and are called protein carbonyls.
Furthermore, levels of thiobarbituric acid reactive substances(TBARS) were measured as a biomarker of lipid peroxidation as shown in Fig. 2 B. Significantly increased MDA was observed intreated cells as compared to untreated control. It has been well established that during normal metabolism, polyunsaturated fatty acids are oxidized and MDA are produced as a byproduct. During pathological conditions, however, the process of lipid peroxidation becomes accelerated resulting in increased generation of reactive carbonyls such as MDA and 4-hydroxynonenal(HNE) [17 , 80] . Protein carbonyl content and MDA levels are,therefore, used as indices to measure oxidative damage hence,serve as potential marker for protein and lipid peroxidation,respectively [81] .
In conclusion, our study supports the theory that intensity of oxidative stress has an imperative role in the tumor suppression as well as carcinogenesis. Increased redox state of the cell is responsible for plethora of cellular functions that mediates cell death. Our findings demonstrate thatinduced oxidative stress has inhibiting effects on cancer cell proliferation. The up-regulation of GSTP1 and Prx2 induced by oxidative stress might play crucial roles in tumor suppression by reducing antioxidant defense system that in turn increases protein oxidation and lipid peroxidation. The interactions between lipid peroxidation, GSH, GSTP1 and Prx2 may modify the role of protein carbonyls to induce tumor suppression.Our study suggests that GSTP1 and Prx2 could be possible therapeutic targets for HCC.
Acknowledgments
We thank Dr. Charles Rice (The Rockefeller University, NY, USA)for providing Huh-7 cells for this study.
CRediT authorship contribution statement
Zehra Hashim:Conceptualization, Data curation, Funding acquisition, Investigation, Writing - original draft.Amber Ilyas:Investigation, Writing - original draft.Shamshad Zarina:Writing -review & editing.
Funding
This study was supported by a grant from the Higher Education Commission, Islamabad - Pakistan (NRPU: 20-4386).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Transjugular intrahepatic portosystemic shunt for a patient with chylothorax in cryptogenic/metabolic cirrhosis
- Differential methylation landscape of pancreatic ductal adenocarcinoma and its precancerous lesions
- Hepatobiliary&Pancreatic Diseases International
- MicroRNAs and long non-coding RNAs in liver surgery: Diagnostic and therapeutic merits
- Alpha-fetoprotein and 18 F-FDG standard uptake value predict tumor recurrence after liver transplantation for hepatocellular carcinoma with portal vein tumor thrombosis: Preliminary experience
- Translationally controlled tumor protein exerts a proinflammatory role in acute rejection after liver transplantation