Differential methylation landscape of pancreatic ductal adenocarcinoma and its precancerous lesions
2020-07-07AksBrriSuhnkrDySumitGultiSupriyoGhtkShijyotiGhoshSupBnrjNiljSikr
Aks h Brri Suhnkr Dy Sumit Gulti Supriyo Ghtk Shijyoti Ghosh Sup Bnrj Nilj Sikr ∗
a Human Genetics Unit, Indian Statistical Institute, Kolkata, India
b Department of Zoology, New Alipore College, University of Calcutta, Kolkata, India
c Department of Gastroenterological Surgery, Calcutta Medical Research Institute, Kolkata, India
d Department of General Surgery, Medical College and Hospital, Kolkata, India
e Department of Gastrointestinal Surgery, Tata Medical Center, Rajarhat, Kolkata, India
Keywords:Pancreatic cancer Methylation CpG islands Epigenetic Biomarkers Precancerous lesions
A B S T R A C T Background: Pancreatic cancer is one of the most lethal diseases with an incidence almost equal to the mortality. In addition to having genetic causes, cancer can also be considered an epigenetic disease. DNA methylation is the premier epigenetic modification and patterns of aberrant DNA methylation are recognized to be a common hallmark of human tumor. In the multistage carcinogenesis of pancreas starting from precancerous lesions to pancreatic ductal adenocarcinoma (PDAC), the epigenetic changes play a significant role.Data sources: Relevant studies for this review were derived via an extensive literature search in PubMed via using various keywords such as pancreatic ductal adenocarcinoma, precancerous lesions, methylation profile, epigenetic biomarkers that are relevant directly or closely associated with the concerned area of our interest. The literature search was intensively done considering a time frame of 20 years (1998-2018).Result: In this review we have highlighted the hypermethylation and hypomethylation of the precancerous PDAC lesions (pancreatic intra-epithelial neoplasia, intraductal papillary mucinous neoplasm, mucinous cystic neoplasm and chronic pancreatitis) and PDAC along with the potential biomarkers. We have also achieved the early epigenetic driver that leads to progression from precancerous lesions to PDAC.A bunch of epigenetic driver genes leads to progression of precancerous lesions to PDAC ( ppENK, APC,p14/5/16/17, hMLH1 and MGMT ) are also documented. We summarized the importance of these observations in therapeutics and diagnosis of PDAC hence identifying the potential use of epigenetic biomarkers in epigenetic targeted therapy. Epigenetic inactivation occurs by hypermethylation of CpG islands in the promoter regions of tumor suppressor genes. We listed all hyper- and hypomethylation of CpG islands of several genes in PDAC including its precancerous lesions.Conclusions: The concept of the review would help to understand their biological effects, and to determine whether they may be successfully combined with other epigenetic drugs. However, we need to continue our research to develop more specific DNA-demethylating agents, which are the targets for hypermethylated CpG methylation sites.
Introduction
Pancreatic cancer (PanCa) is one of the most fatal malignant neoplasm around the world. Nowadays, PanCa is leading the 4th for cause of cancer mortality in USA and is estimated to be the 2nd leading cause for cancer related death by 2030 [1] ( https://seer.cancer.gov/ ). Majority of PanCa is exocrine in origin that develops from ductal cell and is known as pancreatic ductal adenocarcinomas (PDAC) [2] . PDAC is the most common form of PanCa that fills up more than 80% of cases. The aggressiveness of this malignant tumor is due to its asymptomatic feature and is usually detected only when it has already spread to surrounding tissues [3] .It is presumed that PDAC originated from the epithelium of ductal system, because of their ductal phenotype. It has been observed that hyperplastic and metaplastic changes of duct epithelium are commonly associated with ductal adenocarcinoma [3] . The clinical outcome of PDAC is high short-term mortality than any other neoplasm. As per the updated epidemiological scenario, PDAC remains the 4th leading cause of cancer related deaths. In 2018, 55,440 new cases of PanCa were registered in USA and 44,330 individuals died of this disease (SEERS; https://seer.cancer.gov/ ). According to GLOBOCAN 2012 data, the age standardized rate (ASR) of PDAC is 4.9 and 3.6 per 10 0,0 0 0 in men and women, respectively. According to the SEER data 2006-2012 the 5-year survival rate for PDAC is 7.7% [2] . The incidence of PDAC in India is 0.5-2.4 per 10 0,0 0 0 men and 0.2-1.8 per 10 0,0 0 0 women [4 , 5] . The development of PDAC occurs over an extended period of time and mostly it follows a stepwise progression which means transition of a normal pancreatic duct to a pre-invasive precursor lesions namely pancreatic intra-epithelial neoplasia (PanIN), mucinous cystic neoplasm (MCN), and intraductal papillary mucinous neoplasm(IPMN) [6] . Pancreatic ductal lesions are found at various frequencies in the pancreatic parenchyma under different conditions in the pancreas specimens bearing ductal carcinomas, in tumor free pancreas specimens with chronic pancreatitis, and even disease free normal pancreases [7] .
Several factors have been identified as risk factors for PDAC.Smoking and diabetes mellitus have the highest impact on PDAC development. Besides those primary risk factors, obesity, alcohol,increased oil fat consumption, coffee intake, and decreased physical activity are significantly associated with increased risk of PDAC.Supportively, PDAC progression may be attributable to occupational exposures, and this includes chlorinated hydrocarbons, organochlorines, ionization radiation, airborne particles and nitroamines [8 , 9] .The food habit is not the only risk factor but the family history also plays an important role in the progression of PDAC. PanCa, in near family history, is very much associated with the occurrence of PanCa in the descendants. It increases the chance of occurrence of PanCa 2.5 to 5.3 times higher in an individual than those having no such family records [10] . Other risk factors that are closely associated with the progression of PDAC include chronic pancreatitis, allergies, periodontal diseases, several infections likeHelicobacterpylori, cholelithiasis and cholecystectomy, pernicious anemia, hepatitis-B virus (HBV), non “O”blood types and non-steroid anti-inflammatory drugs (NSAIDs) [11] . Pancreatic ductal adenocarcinomas are also highly associated with several known host factors in addition to advanced age, inflammatory bowel disease, sclerosing choledochal cysts, and intra-hepatic or common bile duct stones [3] . Several genetic and epigenetic alterations are involved in development of PDAC and its previous lesions. Activations ofKRASand inactivation ofP16/CDKN2A,SMAD4/DPC4are commonly found in all types of precancerous lesions. Besides this, telomere shortening in PanIN, mutation inPIK3CA,BRAFandGNASin IPMN andRNAF43mutation in MCN are considered to be the major genetic events.TP53is a tumor suppressor gene (TSG) encodes for TP53 protein that regulates cell-cycles particularly through G2/M check point and loss of function of these genes has been identified in both IPMN and MCN [1 , 12] . Sequencing of more than 20,0 0 0 protein coding genes of several hundreds of PDAC demonstrated several genomic alterations including non-silent mutations, amplifications and homozygous deletions in each PDAC tissue sample [13] . These mutations were associated with 12 core signaling pathways. Based on the frequency of somatically altered genes in PanCa, a genetic topographic map of the PanCa can be generated,in which high frequency driver genes are called as “mountains”involvingKRASoncogene,CDKN2A/p16,SMAD4/DPC4,andTP53.The low frequency driver genes named “hills”involveSMARC4A,CDH1,EPHA3,FBXW7,EGFR,IDH1andNF1[14 , 15] . In addition, PDAC patient samples have been shown to contain many chromosomal rearrangements, including gene deletions and amplifications.
Cell carries out functional instructions via various environmental and cellular autonomous processes by synthesizing regulatory non-coding RNAs along with marking DNA and proteins with specific post-translational modifications. This is the base mechanism of epigenetic study. Modifications are referred to as “marks”, which are deposited by “writers”, successively hydrolyzed and degraded by “erasers”and finally recognized and bound by “readers”. The state in which gene expression patterns are connected with their successive phenotypic characteristics became known as “epigenetic landscapes”[16] . In association with genetic changes, another particular area of research focuses around the epigenetic modulation, which plays a vital role leading to disease development, sustainability and progression. Epigenetic alterations correlated with cancer development include silencing of TSG via DNA methylation, inactivation of DNA repair genes by histone acetylation, and miRNA (miR) regulation of cell proliferation, differentiation and apoptosis [17] . The actual definition of epigenetic is alteration in gene expression that is not accompanied by changes in DNA sequences. The main categories of epigenetic alteration that can lead to progression of PDAC are DNA methylation. DNA methylation is very closely related with CpG islands. Approximately 60% of human genes are associated with CpG islands [18 , 19] . However, it is clear that there are normal tissue specific patterns of CpG island methylation and some CpG islands are prone to progressive methylation during aging and development of certain diseases such as PanCa [15 , 20] . DNA methylations occur due to the covalent addition of a methyl group to the 5 ′ position of cytosine and this modification is maintained by a family of gene that is DNA methyltransferases (DNMTs). DNA methylation is categorized into two groups that are hypermethylation and hypomethylation [20 -23] .DNA hypermethylation of other genes except TSG has also been strongly linked with cancer initiation and progression [18] . DNA hypomethylation was observed in cancer cells in many genomic locations such as repetitive sequences, retrotransposons and introns resulting in genomic instability. In the repetitive sequences, a higher rate of chromosomal rearrangements and a higher probability of translocations were observed at retrotransposons [19] . In the last two decades, the field of cancer epigenetics has attracted more interest among basic and clinical researchers, especially after the introduction of specific methods utilized for DNA methylation analysis, including methylation specific PCR (MSP), array methodology,and next-generation sequencing (NGS) after bisulphite treatment,methylC-seq, ChIP-chip and ChIP-seq [19 , 23] . It has been observed that PanIN, IPMN, and MCN have a higher frequency of developing into invasive PDAC by several genetic and epigenetic changes in the tissue environment [24] . Observation of aberrant methylation in PanCa patient samples leads to further investigation of methylation patterns in precursor lesions which finally leads to detection of genes commonly methylated in PDAC patients [25] .
Timely relevance of this systematic review is to summarize all the differential methylation of certain specific genes in several precancerous lesions of PDAC. The epigenetic alterations which occur during PDAC have also been discussed in this review. We deliver the message that, specific epigenetic driver genes drive ductal invasive carcinoma from different kinds of early stages to PDAC. This review provides brief insight into the early epigenetic biomarkers from different biological specimen and therapeutic approaches that specifically target the epigenetic alterations in precancerous lesions and PDAC. The epigenetic modifications that were observed mutually in precancerous lesions and during its progression towards PDAC are also documented. We also highlighted the worldwide incidence and prevalence of PDAC along with the scenario of Indian Subcontinent.
DNA methylation of precancerous lesions of PDAC
A systemic approach occurs in case of PDAC during which a conversion of normal ductal epithelium into precursor lesions occurs, further proceeding towards development of PDAC. Three subtypes of precursor lesions are PanIN, MCN and IPMN [25] . It hasbeen observed that PanIN has a higher frequency of developing into invasive cancer. Observation of aberrant methylation in PanCa patient samples lead to further investigation of methylation patterns in precursor lesions which finally lead to detection of genes commonly methylated in PDAC patients [26 , 27] . Aberrant methylation expression of IPMN was compared with normal pancreatic ductal epithelial cells. Partial methylation ofCDKN1Cpromoter CpG islands along with reduced expression of the protein product was observed in pancreatic cancer cell lines as well as in IPMN. Apart from this, the other hypermethylated genes observed in precursor lesions areppENK,CDKN2A,SPARC,SFRP1,TSLC1,RELN,TFPI2,CLDN5andUCHL1in IPMNs. The epigenetically inactivated genes wereSFRP1,RPRM,SPARC,CLDN5,LHX1,NPTX2,ppENK,andCDKN2Ain PanINs, whereasCDKN2Ain MCN. From the previous data the evidences support the fact that aberrant hypermethylation of CpG islands occurs in the precursor lesions and further progresses towards PDAC [26 , 27] . Sato et al. [26] compared global gene expression profiles between IPMN and normal pancreatic ductal epithelial cells and found thatCDKN1Cwas under expressed gene in IPMN.Partial methylation ofCDKN1Cpromoter CpG islands along with reduced expression of the protein product was observed in pancreatic cancer cell lines as well as in IPMN. These evidences support the fact that aberrant hypermethylation of CpG islands occurs in the precursor lesions and then further progress towards PDAC( Table 1 ) [28] .

Table 1.Hypermethylation genes in precancerous lesions of pancreatic ductal adenocarcinoma.
AberrantmethylationinPanINs
An extensive study on methylation of different genes in PanIN and PDAC had been performed by Fukushima et al. [27] . They reported two genes, preproenkephalin (ppENK) andp16, that are found aberrantly get methylated in both PDAC and PanIN [27] .In this study, the amplified DNA from 102 PanINs and 99 PanINs were analyzed to observe the aberrant methylation in CpG island ofppENKandp16,respectively. This study found that the prevalence of methylation in CpG islands ofppENKwas increased with increase of histological grades of PanIN. Strikingly, aberrant methylation ofppENKwas observed in 7.7% (2 of 26) patients with PanIN-1A, 7.3% (3 of 41) with PanIN-1B, 22.7% (5 of 22)with PanIN-2, and 46.2% (6 of 13) with PanIN-3. This result clearly indicated thatppENKmethylation was more frequently present in high-grade PanINs (PanIN-3) than in low-grade PanINs(PanIN-1 and PanIN-2) (P= 0.005) [27] . In opposite, only 8 of 99 PanINs with different grades had shown methylation ofp16CpG islands. These include 12.0% (3 of 25) PanIN-1A, 2.6% (1 of 38) PanIN-1B, 4.5% (1 of 22) PanIN-2 and 21.4% (3 of 14)PanIN-3. Althoughp16methylation was seen more frequently in high-grade PanINs than it was in low-grade PanINs, this difference was not statistically significant (P= 0.146) [27 , 29] . A comparative study on aberrant methylation ofppENKandp16had been performed in three different groups of patients with PDAC, other neoplasms, and chronic pancreatitis. This was done to investigate whether they have any role in development of PanCa by possessive methylation of PanIN or not [30 , 31] . Methylation specific PCR analysis in PanINs from 60 patients associated with PDAC had shown a prevalence ofppENKCpG island methylation in PanINs and it tended to increase with histological grades (in PanIN-1A,PanIN-1B, PanIN-2, and PanIN-3; 22.2%, 12.0%, 35.7%, and 50%,respectively). DNA amplification of 58 PanINs micro dissected from pancreas with an invasive ductal adenocarcinoma (IDA) were examined forp16and seven of the lesions showed methylation ofp16CpG islands with increasing rate in high grades [20.0% (2 of 10) PanIN-1A, 4.8% (1 of 21) PanIN-1B, 7.1% (1 of 14) PanIN-2 and 23.1% (3 of 13) PanIN-3]. With respect to the study exploring this group of PanINs,ppENKmethylation was more frequently seen in high-grade PanINs than in low-grade PanINs (P= 0.001) and there was no significant difference in the frequency ofp16methylation(P= 0.368). The oligonucleotide microarray analysis of invasive pancreatic cancer cells after treatment with DNA methyltransferase inhibitor 5-aza-2 ′ -deoxycytidine (5Aza-dC) has identified a large panel of 475 genes [26] . In the midst of this significantly large gene panel, eight genes are considered as most potential agent in invasive PanCa, which have shown hypermethylation in cancer cell but completely unmethylated in normal cell. The explored genes were cadherin 3 (CDH3),RPRM,LHX1,NTPX2, claudin 5 (CLDN5),SARP2,SPARC, andST14[26] . It has been reported that at least one of the above mentioned eight genes was found aberrantly methylated in different grades of PanIN lesions (71%, 57%, 60%and 92% in PanIN-1A, PanIN-1B, PanIN-2, and PanIN-3, respectively) [26] . A study on CpG islands methylation with DNA from 65 PanIN lesions (17 of PanIN-1A, 21 of PanIN-1B, 15 of PanIN-2,and 12 of PanIN-3) was performed by utilizing MSP. The result showed differential CpG island methylation at each locus of above mentioned eight genes (13% forCDH3, 30% forReprimo, 14% forLHX1, 20% forNPTX2, 13% forCLDN5, 23% forSARP2, 28% forSPARC,and 0% forST14) [26] .
The study by Jansen et al. [32] suggested that various genes whose inactivation by methylation depend on the stages, the timing of methylation in neoplastic precursor lesions depends on the function of the gene, rather than on global methylation patterns.They also observed that methylation ofp16andppENKis also uncommon in early PanINs compared to late PanINs and in PDAC( Table 1 ) [32] . The geneTSLC1was a recently identified TSG located on chromosome 11q23.2. The MSP study after bisulfite treatment of genomic DNA of pancreatic cancer cell line had shown aberrant methylation in CpG islands in 24% of cases, whereas no alternative methylation had been observed in normal pancreas cell line. Further examination of PanIN lesion for observation ofTSLC1methylation has revealed that, in 29% cases of PanIN-3 aberrantly methylated at 5 ′ CpG region was found and its expression is repressed,which disabled the tumor-suppressor activity; but none was found in PanIN-1 and PanIN-2 lesions ( Table 1 ) [32] .
AberrantmethylationinIPMNs
A predominantly acknowledged cystic neoplasm of the pancreas is the IPMN that has a potentially vast array of malignancy and has been reported as a precursor to invasive ductal adenocarcinoma [33] . The information from literature surveys have shown that aberrant methylation of the promoter CpG island is a common mechanism associated with the silencing of TSG which was also frequently observed in IPMNs [34 -36] .CDKN1C/p57KIP2,ppENK,p16,thrombospondin1,CyclinD2,SOCS-1,RELNandTFPI2genes have been identified which were found aberrantly methylated in IPMNs [34 , 37 -40] . Amongst these mentioned genesppENKandp16are more frequently methylated in high grade of IPMNs than low grade of IPMNs (ppENK, 82% vs. 28%,P= 0.0 0 02;p16, 21% vs. 0%,P= 0.04) [34] . A report by Hong et al. demonstrated an increase of hypermethylation in IPMN (e.g.,p16/CDKN2A,CDKN1C,SRYandSOX17) [39] . An increasing number of hypermethylated loci are associated with an increasing grade of dysplasia ( Table 1 ).
Martin et al. [41] have identified human hedgehog interacting protein (HHIP) as a target of aberrant methylation in PanCa. Specifically, they find that theHHIPpromoter is hypermethylated in approximately half of all PanCa [41] . Their observed results showed that DNA methylation of theHHIPpromoter plays an important role inHHIPinactivation. The above study also demonstrated the differences in expression and methylation pattern ofHHIPbetween the cell lines and in the IPMNs, andHHIPmethylation may be an important factor for progression of PanCa ( Table 1 ) [41] .
AberrantmethylationinMCN
Cystic tumors of the pancreas are relatively uncommon, accounting for less than 5% of pancreas exocrine tumors. There are two types of tumors found, MCN and serous microcystic adenoma(SMA) [42] . The MCN are classified as benign, borderline, carcinoma in situ, and invasive carcinoma based on the severity of dysplasia of the lining epithelium or presence of stromal invasion.Few studies have been performed on epigenetic alteration in MCN,and the knowledge about aberrant methylation in different candidate genes in development of MCN and its prognosis into PanCa is very obscure. A study with a small sample size of 14, have shown that aberrant methylation ofp16andp14occurred in 14% (2 of 14) cases of MCN. Methylation specific PCR analysis ofVHLwas performed but 0% occurrence found in MCN, whereas 7% cases recorded for SMA ( Table 1 ) [42] .
Aberrantmethylationinchronicpancreatitis
Significant relevance pinpointing at pancreas’s chronic, inflammatory process pursued by phenotypic changes of irreversible nature is defined as chronic pancreatitis. Main reason of high mortality rate in PanCa is relative lack of symptoms early in diseases process, resistant to many treatments and the hidden location of pancreas. Many of these symptoms, such as pain, diarrhea, weight loss, jaundice, and mal-absorption, are similar to chronic pancreatitis. From chronic pancreatitis to diagnosed PanCa needs at least 5 years [43] . Epidemiological studies have shown that chronic pancreatitis is one of the major risk factor for PanCa, with a 2.3 to 18.5 fold increased risk compared to normal controls; numerous extrinsic and intrinsic pathways have been identified to be involved in the connection between chronic pancreatitis and PDAC. Several deregulated signaling pathways, such as cytokines, NF-κβ, reactive oxygen species (ROS) and PPARγ, have been identified in PDAC and chronic pancreatitis [44] . Matsubayashi et al. [45] tested 17 genes of pancreatic juice from patients with PanCa, chronic pancreatitis and controls and found that the percentage of genes methylated in patients with PanCa was higher than that in those with chronic pancreatitis and controls but no particular gene was useful for the diagnosis of chronic pancreatitis in comparison with controls. When comparing chronic pancreatitis and PanCa, 14 genes were selected and 9 of the 14 gene promoters were specific to chronic pancreatitis that include Cyclin D2 (CCND2) (23%),ESR promA(22%),hMLH1(18%),CDKNIC(27%),MYOD1(26%),SYK(23%),PRproximal promoter (22%),Mucin2oligomericmucus(17%) andRb1(16%) ( Table 1 ) [45] .
Hypermethylation in PDAC
Pancreatic ductal adenocarcinoma progression via abrogation of various tumor suppressor pathways has been relatively well documented in the progression of PanCa. Nowadays, it is apparent that epigenetic alteration such as main methylation and the transcriptional changes of affected genes plays an important role for progression of various human cancers from its precancerous state to PDAC. Two forms of frequently observed methylation changes in cancer are global hypomethylation in repetitive position and gene specific hypermethylation of CpG islands [46 , 47] . As it has been mentioned above, DNA hypermethylation has been a major driving force in the progression of PDAC [31] . CpG islands are usually targeted as the regions with maximal hypermethylation due to the presence of high amount of CpG dinucleotide in the promoter region of the DNA. Anomaly methylation of promoter CpG islands has been associated with gene silencing along with TSG inactivation. The TSG and CpG islands are unmethylated in healthy situations and act as potential targets for DNA methylation in PDAC patients. Ueki et al. [48] emphasized on aberrant CpG island methylation. At least one out of 12 cancer related genes (CRG)was observed in 60% cases of carcinomas. Promoter methylations were observed in genes likep16,RARβ,CACNA1G,TIMP-3,THBS1,hMLH1andDBC1with frequency varying from ~5 to 20%. Loss of expression is the outcome of the above epigenetic changes. Higher rate of methylation was observed in 4 CpG islands namelyMINT1,−2,−31, and −32(11% −73%). Previous observations also stated thathMLH1methylation outside the promoter region inactivates transcription. During normal situationhMLH1stays methylated but in some cases the rate increases to inactivate its expression [48] .In case of PDAC there were several CRGs that are hypermethylated in their promoter regions likeSPRAC,p16,RASSF1A,3-0ST-2,Cyclin D2,SOCS-1,RAR-b,APC,RELN,TFPI-2andppENK, which play an important role for the progression of disease. DNA hypomethylation also occurs at the 5 ′ region of certain genes in PanCa. For example,S100A4was associated with hypomethylation at specific CpG site [49] . It has been exhibited thatRASSF2A,one of TSGs,is inactivated by promoter hypermethylation. The inactivation ofRASSF1Amay rule out the necessity of K-ras activation to alter Ras signaling in PanCa [50] . The methylation status ofRASSF2Awas screened by Zhao et al. in 8 human pancreatic cancer cell lines and 41 paraffin embedded tissues of PDAC samples where they found 100% hypermethylation in pancreatic carcinoma cell line and 22% hypermethylation in paraffin embedded tissues [51] .Omura et al. used genome-wide profiling and concluded that aberrant methylation in PanCa cells targeted towards the promoter along with the CpG islands. They identified hypermethylation inMDFI,has-miR-9-1,ZNF415,CNTNAP2andELOVL4(96%, 89%,86%, 82% and 66%, respectively) of PanCa [52] . In a study to characterize DNA methylation patterns, Tan et al. showed the methylation pattern of 807 genes and identified 23 candidate genes with hypermethylation on CpG islands in PanCa. According to them, the genes of tyrosine kinase family likeKIT,FLT1,FLT3,NTRK3andEPHA5were also hypermethylated and as a result they contributed to the resistance of certain tyrosine kinase drugs in PanCa due to under expression [53] . Some candidate genes that caused PDAC through hypermethylation wereLIG3,MEST,SFN,VAV2andLIf[53] . Syren et al. analyzed PDAC sample tissues and cell lines and found that PanCa displayed hypermethylation of promoter genes namelyBNIP3,ELOVL4,ZNF415,MDFI,CNTNAP2,SPARC/osteonectin,ppENK,DBC1,FLT1andEYA[54] . Matsubayashi et al. analyzed pancreatic juice collected from 155 individuals with pancreatic disease and identified 118 methylated genes out of 187 genes. The pancreatic juice samples of PanCa patient contain more methylated genes as compared to juice of patients without PanCa [45] . Sato et al. found that aberrant hypermethylation ofTFPI-2gene’s promoter CpG island is associated with its loss of expression. Hence, they concluded that the epigenetic inactivation ofTFPI-2gene is a contributing factor in PDAC aggressive phenotype( Table 2 ) [40] .
The aberrant hypermethylation in promoter of certain cancer related genes can cause carcinogenesis by up-regulating or downregulating the cell cycle genes, DNA repair genes and other signaling pathway [43] . In PanCa several CRG has been reported to be hypermethylated which becomes the lead cause of carcinogenesis. Cancer related genes likep16,RASSF,3-OST-2,Cyclin-D1,SOC-1,RAR-β,DBC1andAPChave become inactive after promoter hypermethylation. Another TSG,RASSF1Awas reported in PanCa which after promoter hypermethylation becomes silent. Promoter hypermethylation can be detected using MSP as previously mentioned.This is a very advanced and sensitive technique with the ability to detect very small number of cancer cells in peripheral blood. Another diagnostic marker cell free DNA promoter hypermethylation for PDAC has the ability to differentiate between malignant and benign lesions. Xu et al. found that the hypermethylation of the control region ofDUSP6is the reason behind its undoing nature in human PanCa cells [55] .
Sato et al. [56] demonstrated that DNA methylation influencesCXCR4expression in human PanCa. Again, they used MSP, combined with bisulfite restriction analysis, and bisulfite sequencing,and found that the 5 ′ CpG islands of theCXCR4gene to be unmethylated in normal pancreas, whereas promoter hypermethylation was detected in 45% (9 of 20) of pancreatic cancer cell lines and in 46% (46 of 100) of primary PDAC [56] . Yi et al. observed that theBNC1hypermethylation have a sensitivity of 79% and a specificity of 89% in PanCa [57] . Several hypermethylated genes, such as Cadherin (CDH13),HIC1,HIC2,HOXA9andIGFBP5are correlated with various types of cancers ( Table 2 ) [57] .
Hagihara et al. [58] used methylation-sensitive-representational difference analysis (MS-RDA) to isolate 111 DNA fragments derived from CpG islands and found that 35 of them were in the 5 ′ regions of known genes. They also analyzed MSP of the 5 ′ CpG islands in seven pancreatic cancer cell lines and two pancreatic ductal epithelial cell lines and found that 13 genes (RASGRF2,ADAM23,NEF3,NKX2-8,HAND1,EGR4,PRG2,FBN2,CDH2,TLL1,NPTX1,NTSR1andTHBD) were silenced by methylation of their 5 ′ CpG islands in promoter region. MSP of 24 primary PanCa showed that all these genes, except forTHBD, were methylated in at least one cancer including PanCa. Some of those genes were suggested to be potentially involved in PanCa development and progression( Table 2 ) [58] .
Differential methylation identified in PDAC by NGS studies
The identification of genome wide pattern of DNA methylation is necessary to detect epigenetic dysregulation in PDAC. The highdensity array was used by Nones et al. to capture the methylation profile of 167 untreated resected PDAC. They compared PDAC samples to 29 adjacent untransformed pancreatic tissues and identified 3522 aberrantly methylated genes [59] . The samples were collected as part of Australian Pancreatic Cancer Genome Initiative (APCGI).
As documented in a review by Iguchi et al. [60] , hypermethylation in the genesROBO1,ROBO3,SLIT3andSLIT2play an important role in PDAC. The epigenetic perturbation of axon guidance pathways was analyzed by using bisulfite amplicon deep sequencing and qRT-PCR and suppression ofSLIT-ROBOsignaling was observed [59-62] .
Hypomethylation in PDAC
DNA hypomethylation was the initial epigenetic abnormality recognized in human tumors. Repeated DNA elements were adjacently connected with hypomethylation of DNA in cancer biology, irrespective of that malignancy associated DNA hypomethylation has still received very minute recognition. Hypomethylation in highly repeated DNA sequences was only considered as reason for cancer. However a hypomethylation transcription regulatory region in cancer has been recognized as an important epigenetic abnormality after the discovery of high resolution genome wide studies. In PDAC, aberrant loss of methylation is detected at genomic level as well as sequence specific level. The loss of methylation leads to many genetic alterations and instability. It was discovered that the deficiency in folate and vitamin B 12 may contribute to hypomethylation by decreasing the level of methyl group donor Sadenosyl methionine (SAM) which resulted in decreased biosynthesis of thymidine from uracil [14 , 19 , 63] . The cancer developmentis also caused by misplacement of uracil into thymidine which not only imbalance the nucleotide pool but also increase DNA strand breaks. Methylene tetra hydro folate reductase (MTHFR) gene is very important for the PDAC prospective because defectiveMTHFRgene presents in PanCa that causes more DNA hypomethylation, resulting in increased chromosomal losses. Unlike hypermethylation,hypomethylation causes the over-expression of protein and genes.For example, in PanCa the DNA hypomethylation at 5 ′ regions of certain gene andS100A4linked hypomethylation at specific CpG site within the first intron also associated with overexpression of encoded proteins [49 , 64] . Sato et al. reported 19 out of 32 genes had methylation and 7 of them (CLDN4,LCN2,TFF2,MSLN,S100A4,SFN/14-3-3σ,andPSCA) were hypomethylated in pancreatic cancer cell line as well as primary PanCa [65] . The analysis of methylation with oligonucleotide array identified two genes includingS100PandMASPwhich are aberrantly hypomethylated. Rosty et al. analyzed National Center for Biotechnology Information (NCBI) Serial Analysis of Gene Expression (SAGE) database and found thatS100A4was expressed in 95% pancreatic carcinoma cell line, and 11 out of 12 pancreatic cancer cell lines had hypomethylation at specific CpG site within the first intron ofS100A4(100%) gene( Table 3 ) [49] . Tan et al. identified 35 candidate genes with hypomethylation among 807 genes in PanCa [53] . In several studies conducted by multiple research groups, it has been found thatCCND1andCCND3 genes were over expressed due to hypomethylation in PanCa [66 , 67] . Mucins likeMUC2andMUC5ACare not expressed in normal pancreas but the promoter methylation causes mucin expression in pancreatic carcinoma cells. About 86% −100%invasive adenocarcinoma expressedMUC5AC[53 , 68] . Zhu et al. detected hypomethylation inMUC4gene at 116 micro dissected foci in 57 patients with PDAC ( Table 3 ) [69] .

Table 2 List of hypermethylation genes in pancreatic ductal adenocarcinoma.
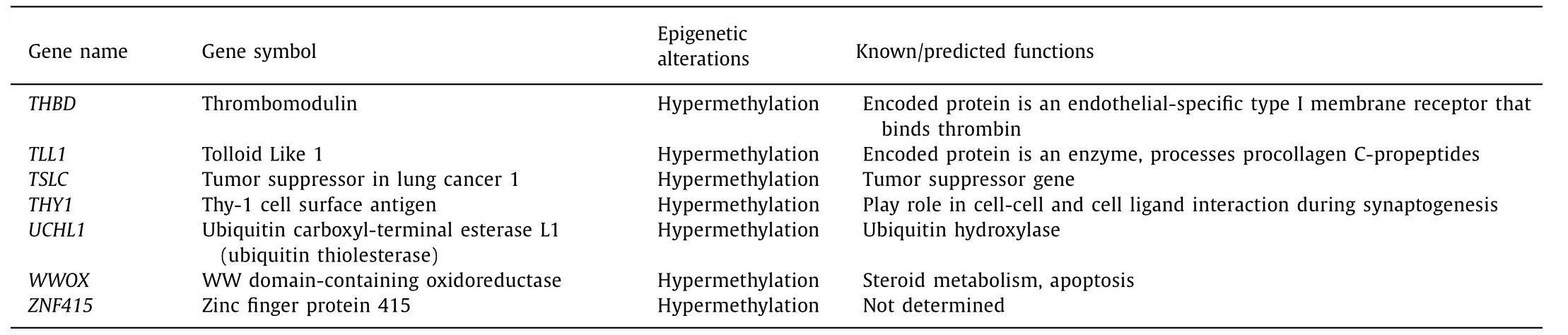
Table 2 ( continued )
Next generation epigenetic study of hypomethylation of PDAC
To identify methylation pattern of genes in PDAC, Vincent et al. [70] performed methylated CpG island amplification followed by CpG island microarray where they analyzed 27,800 CpG islands covering 21 MB of the human genome. The frozen primary PanCa tissues and normal pancreatic tissues were obtained from patients at John Hopkins Hospital and then they analyzed number of genes that are silenced by DNA methylation. The reported genes were TSG genes (STK11andWT1), stem cell pluripotency genes(BM11,BMP3,andFOXD3), cell adhesion genes (CDH2,CDH4,PCDH1andPCDH10)and also genes involved in WNT signaling (WNT5A,WNT7AandWNT9A). By using MCAM (microarray hybridization)assay, they identified many candidate hypomethylated genes in PDAC [70] . Genes responsible for chromatin modification includingSETD8,PRMT1,CTR9,KDM6Aand chromatin assembly proteinHIRIP3were over expressed in PanCa samples as compared to normal ductal epithelium. The Genome wide analysis (GWA) done by Vincent et al. also reported 3 oncogenes (JUNB,MYBandFOS) to be hypomethylated in PanCa ( Table 3 ) [60 , 70] .
Common hypo- and hypermethylated genes between PDAC and its precancerous lesions
There are several methylated genes that were observed in both the precancerous lesions as well as in PDAC. These common methylation patterns observed are the early epigenetic drivers in the progression of precancerous lesions towards PDAC. These genes would have bigger impact on the development and the progression of PDAC from its precancerous stages. Previous studies had reported two genes,ppENKandp16, which are found aberrantly methylated in both PanIN and PDAC. As documented in a review by Omura and Goggins, partial methylation ofCDKN1Cpromoter CpG island was observed in both IPMN and pancreatic cancer cell lines as a reduction in expression [64] . According to a review by Iguchi et al. [60] ,WNK2was silenced during progression of PanIN to PDAC hence contributing in both the cases. It was also observed thatMUC4was overexpressed in both precursor lesions and in PDAC.TheAPCandp16genes are vital in the cell cycle regulation asAPCgene is an extremely important component in theAPC/β-catenin pathway. Thep16also plays a vital role and DNA methylation ofp16leads to silencing of it in PanIN and PDAC, hence highlighting the carcinogenic progression from precancerous lesions to PDAC( Table 4 ) [60] .
Peng et al. found thatAPCandp16genes were the most frequently methylated and the frequency of DNA methylation of these two genes were increased from PanIN to PDAC. Another observation reported was that the down-regulation ofBRCA1gene may be due to DNA methylation and it was observed across chronic pancreatitis to PDAC [71] . Delpu et al. in a review pointed out that 8 genes were differentially methylated and the methylation pattern varies amongst normal epithelia and PanIN lesions [72] . It indicates that in case of PDAC initiation CpG island hypermethylation is an early event hence justifying the involvement of methylation pattern in progression from PanIN to PDAC. The genes reported in this study areCDH3,reprimo,CLDN5,LHX1,NPTX2,SARP2,SPARCandST14.Another subset containingp16,ppENK,p14,p15,p17,APC,hMLH1,E-CadherinandMGMTwere observed in IPMN showing similar results as in PanIN. These aberrant DNA hypermethylation highlights the fact that methylation acts as a driving force from precursor lesion progression to PDAC [72] .WNK2,a cytoplasmic serine/threonine kinase, was hypermethylated in both chronic pancreatitis and PDAC.WNK2silencing supports PDAC progression [60] . Systematic hypomethylation ofMUC4may play a key role in carcinogenesis in PDAC ( Table 4 ) [60 , 69] .
Epigenetic therapeutic and diagnostic importance of methylation marker in PDAC
Most of the DNA methylation in the human genome occurs on the cytosine in the CpG dinucleotides. These high density CpG sites or sequences are often found in promoter region of many genes. PanCa is a disease of epigenetic, as well as genetic ab-normalities. During the development of PDAC epigenetic changes take place in parallel with genetic changes. Pancreatic tumors are often diagnosed in advanced stages of the disease notably when it has become too late for surgery to cure the disease. Difficulty in distinguishing the neoplastic or non-neoplatic nature of a lesion when found in pancreas is the reason that many researchers have sought to find accurate markers of pancreatic neoplasia. Cancer cells may release cfDNA into the blood. The best known epigenetic marker is DNA methylation. Aberrant DNA methylation seems to occur in early-stage tumors, causing loss- and/or gain-of-function of key processes and signaling properties. Detection of aberrant DNA methylation is technically simple, and cost effective. More than 5% of all known genes (13,023) are promoter methylated in an individual. Therefore, detection of aberrant DNA methylation is potentially a good early indicator of existing PanCa and even a risk assessment of future development of PanCa. The current advances in epigenetic biomarkers also act as potential substrates for detection of PDAC. Many glycoproteins and protein encoding genes can also be considered as biomarkers due to their expression of epigenetic changes in promoter regions, hence, having potential use as diagnostic and prognostic biomarkers for detection of PDAC.Recently, there have been multiple successful reports of DNA methylation screening using various body fluids such as stool,sputum, saliva, urine for various cancers. DNA hypermethylation or hypomenthylation can be detected in the cfDNA in plasma and serum and potentially in tumor specific and important for blood-based diagnostic markers for PDAC [73 -76] . Endoscopically collected pancreatic juice from PDAC patients also has the diagnostic use of detecting methylation. Endoscopic techniques like endoscopic pancreatic function testing are significantly helpful in characterizing chronic pancreatitis [77] . However, the studies faced difficulties in differentiating between malignant and benign pan-creatic disease. None of the genes have the potential to serve as an individual diagnostic marker. On course of developing and testing a biomarker for PanCa, inclusion of relevant control groups with benign pancreatic disease is very important to enable differentiation of PanCa specific hyper- and hypomethylation related to pancreatic disease in general. Late diagnosis of PDAC is the reason behind its lethal nature and seeks efforts to obtain proper diagnostic biomarkers for early diagnosis of the disease. In this quest of identifying early diagnostic marker tools, various procedures have been applied like genome-wide DNA methylation patterns which are a novel approach to this cause. Screening pancreatic tumor cell lines along with PDAC tumor samples for methylated genomic areas has been performed via various profiling assays. Even the cfDNA obtained from blood plasma of patients was screened as a potential diagnostic marker. Identification of hyper- and hypomethylated multi-markers panel is the key for PDAC therapy. cfDNA can be found in both healthy and PDAC patients but the amount is higher in the blood plasma in patients with PDAC. Blood sample is used as an important approach to identify the presence of methylation.However, a larger gene panel needs to be studied to get sufficient accuracy. For the diagnosis of PanCa, cfDNA hypermethylation can be served as blood based biomarker. The hypermethylated genes includingBMP3,RASSF1A,MESTv2,SFRP1,SFRP2,TFPI2andAPCwere identified from the patients with PDAC. Chronic pancreatitis and acute pancreatitis were used as nonmalignant control against PDAC. cfDNA hypermethylation hence can successfully distinguish chronic or acute pancreatitis from PDAC with 76% sensitivity and 83% specificity [73] . The most common analytical techniques are high throughput global methylation profiling assays, using bisulphate DNA sequencing and MSP ( Table 5 ) [73 , 74 , 78 , 79] .
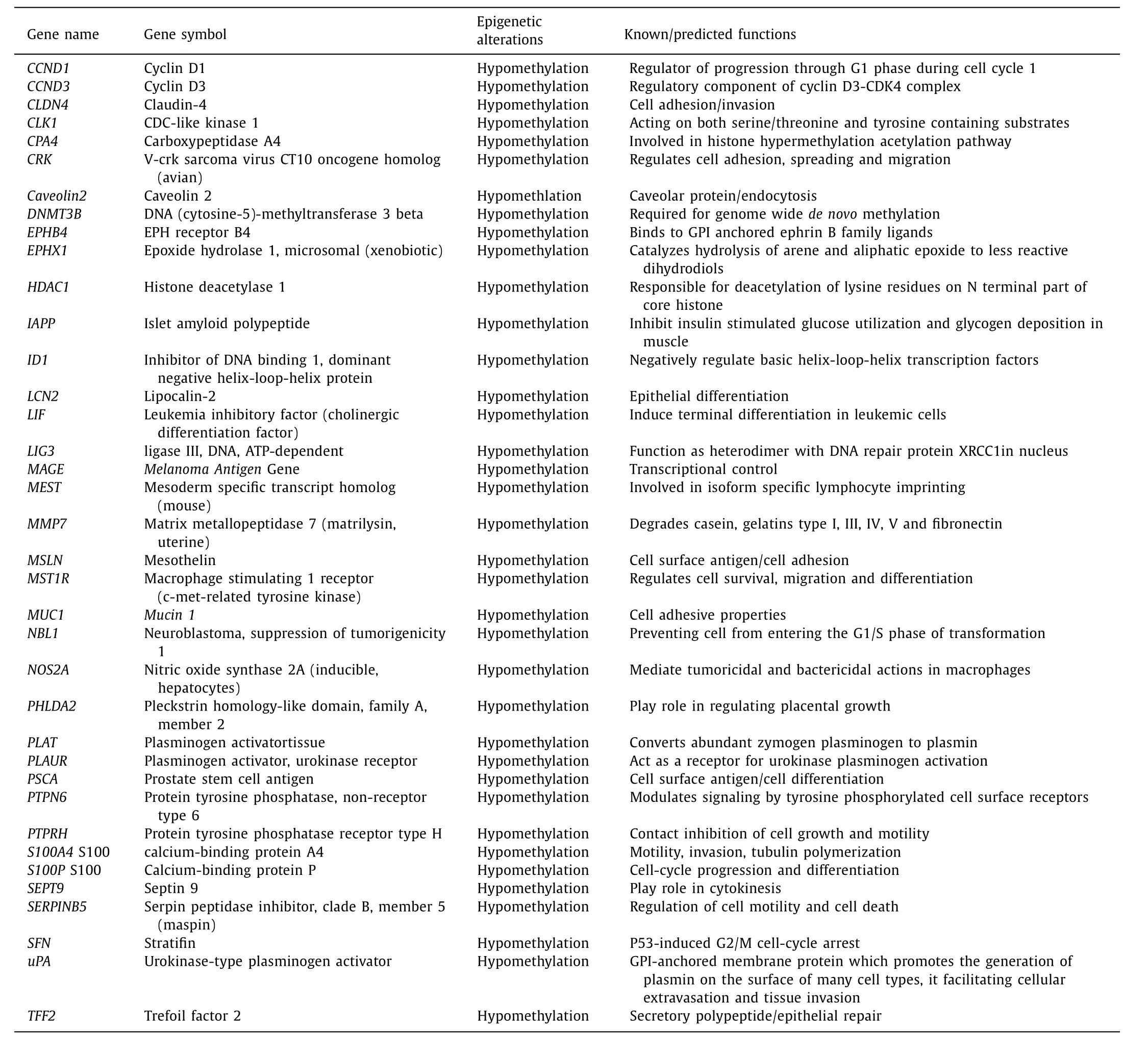
Table 3 List of hypomethylated genes in pancreatic ductal adenocarcinoma.
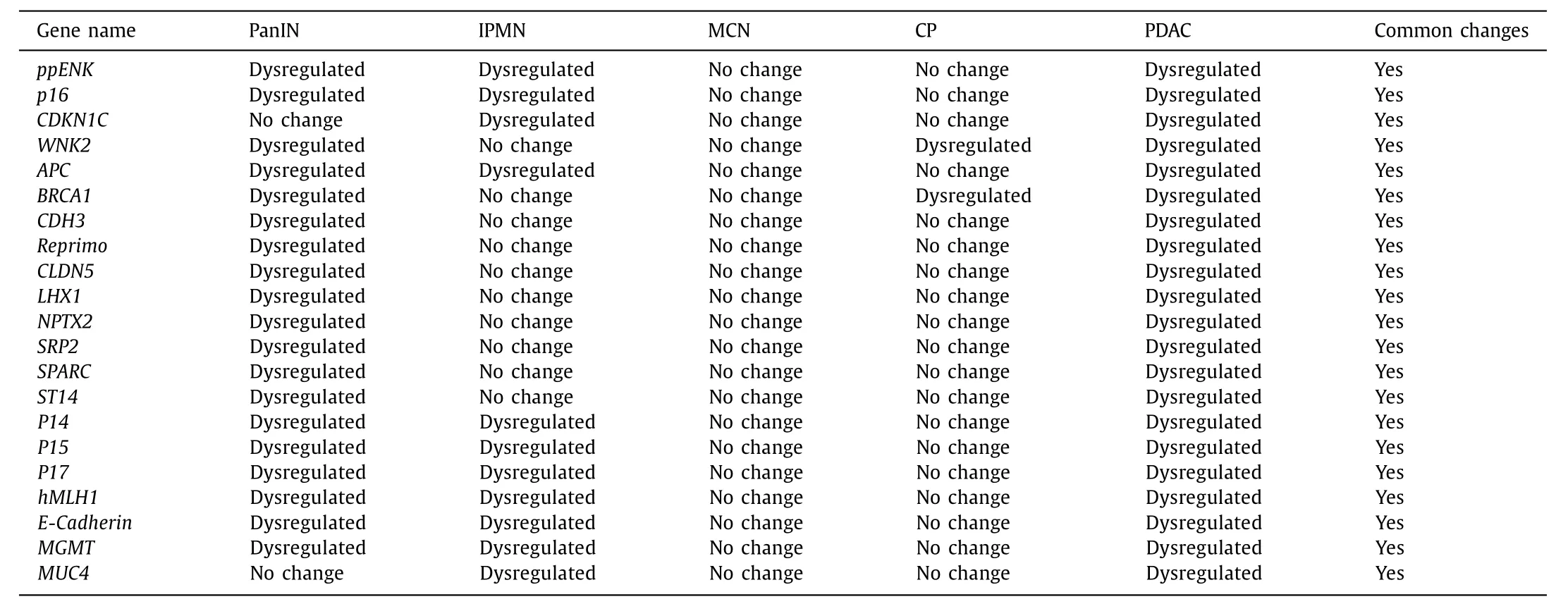
Table 4 Common hypo- and hypermethylated genes in pancreatic ductal adenocarcinoma and its precancerous lesions.
The development of early detection strategy has improved the clinical diagnosis for PanCa. The detection of epigenetic alterations by using new techniques MSP and microarray, DNA methylation profiling assay, colony formation assay, RNAi transfection approach makes the diagnosis easier and simpler.REG4, a member of the regenerating gene (REG) family is a potential biomarker that may link pancreatitis to an earlier diagnostics of PDAC. Although first discovered as a biomarker for colorectal cancer, it has since been found to be associated with PDACs, gastric cancer, relapsed prostate cancer and inflammatory bowel diseases. ThoughREG4is correlated with numerous malignant as well as non-malignant diseases, the specificity is better for PDAC compared to other malignant disease ( Table 5 ) [80] .
From different scientific reports it was observed that the cytosine in CpG dinucleotides is the most common place where the DNA methylation takes place. In the promoter region of many genes, these high density CpG sequences are found to be methylated which is regulated by DNA methyltransferases (DNMTs). The hypermethylation in the promoter region leads to gene silencing whereas hypomethylation leads to overexpression of the gene product. In accordance with the studies carried out by various respective researchers, the pattern of DNA methylation in PanCa was reported where they identified methylation on different CpG sites. Mucin is a type of glycoprotein that remains overexpressed in PDAC and is important for epithelial cell polarity, cell proliferation and differentiation. According to Yokoyama et al., several mucins are suggested as possible prognostic and diagnostic biomarkers, which are interesting and might render other early detection biomarkers [81 , 82] . Hypomethylation occurs in the promoter region ofMUC1,MUC2,MUC3A,MUC4,MUC5ACandMUC17.An aberrant expression ofMUC4and hypomethylation ofMUC4were observed in IPMN and PDAC but not in normal pancreas [54] .Few novel serum biomarkers obtained areIL-10_P348,LCN2_P86,ZAP70_P220,AIM2_P624,TNFRSCF10C,ACIN1,Line-1andALUrepeats.Cell free nucleosome markers for detection of PDAC are5MC,H2AZ,H2A1.1andH3K4Me2[74] . The methylation status of few mucins(MUC1,MUC2andMUC4) showed similar mucin production in both pancreatic fluid and tissues. The mucin methylation status in pancreatic fluid helps in diagnosis of PanCa. The subtyping of IPMN(gastric type vs. intestinal type) aberrant methylation of few potential candidates likeNPTX2,FOXE1,QMSP,TFPI2,p16andCyclin D2were also noted [77] . A panel of 5 ′ CpG sites like interleukin 10 (IL10_P348), lipocalin 2 (LCN2_P86), zeta chain associated kinase (ZAP70_P220), absent in melanoma 2 (AIM2_P624) and T cell acute lymphoblastic leukaemia (TAL1_P817) were measured by Pedersen et al. from the peripheral blood leukocyte DNA of 132 patients with PDAC. It was done as a validation test to yield sensitivity and specificity of 72% and 70%, respectively, [83] . Glycine N-Methyltarnsferase (GNMT), a regulator of methylation processes and of folate metabolism, is frequently hypermethylated in PDAC.The named oncogeneBNC1which is a TSG andADAMTS1, extracellular matrix metalloprotease (EMM) with antiangiogenic func-tion, were observed hypermethylated in the PDAC and PanIN-3.The cell cycle regulatory gene Cyclin D2 (CCND2) which functions in the progression from G1 to S phase of the cell cycle, are found to be hypermethylated in PDAC and IPMNs with respect to normal pancreatic tissues. The promoter regions ofSIP1,ELOVL4,ZNF415,MDFI,CNTNAP2,SPARC/osteonectin, andppENKwere hypermethylated in both pancreatic cancer cell lines and human PDAC tissues. In addition to that, promoter regions ofDBC1,FLT1, andEYA4were found to be hypermethylated in PDAC tissues. After validating the result, they obtained 72% of sensitivity and 70% of specificity [74] . Dauksa et al. [84] investigated methylation levels at other CpG sites in the promoter region of TSG,p16,RARβ,TNFRSF10C,APCACIN1,OST2,BCL2,CD44andDAPK1in the blood of 30 PDAC patients. They also evaluated the methylation pattern ofAlurepeats andLINE-1and observed that the methylation ofLINE-1andAlurepeats were decreased in PDAC patients as compared to healthy controls [74 , 84] . Another geneNPTX2has emerged as a serum methylation marker for PDAC. Park et al. compared the serum methylation levels of theNPTX2gene in 104 PDAC patients and 60 chronic pancreatitis patients and found that methylation levels were significantly higher in PDAC than in chronic pancreatitis [85] . The panel has 28 genes, and 19 genes were found significantly hypermethylated in plasma/serum samples. Henriksen et al.have observedBNC1to be hypermethylated in only 36% of PDAC with a specificity of 94% [73] . Consistent with previous reports, the gene panel did not demonstrate a single gene, which could be used as an individual diagnostic marker for a PanCa. They have developed diagnostic prediction model (age>65 years,BMP3,RASSF1A,BNC1,MESTV2,TFPI2,APC,SFRP1andSFRP2) that can differentiate between PDAC and pancreatic non-tumor tissues. Klett et al. found the following genes,p16,p53,SMAD4,BRCA2,STK11,hMLH1,hCDC4,MKK4,ppENK,SPARC,TFPI2,FOXE1,NPX2,TSLC1,p14,p57andCy-clinD2either hyper- or hypomethylated in PDAC but rarely in nonneoplastic tissues [86] . In a study for early detection of PanCa, Joo et al. used a panel of 8 genes (TFPI2,ASCL2,BNC1,TWIST1,BINP3,ADAMST1,PNMTandEVL)for detection of methylation in promoter region of genes for identifying novel biomarkers in PanCa.The most frequently methylated gene wasBNC1(91%), followed byADAMTS1(67%),ASCL2(65%),BNIP3(49%),TFPI2(54%),EVL(47%),andPNMT(27%) [87] . SerumBNC1andADAMTS1methylation are significantly higher in PDAC patients compared with healthy volunteers. The sensitivity was 79% and 48% forBNC1andADAMTS1,respectively. The specificity of detection was 89% forBNC1and 92%forADAMTS1,respectively, [57] . Matsubayashi et al. analyzed aberrantly methylated DNA from pancreatic juice and found that the quantitative methylation-specific PCR (QMSP) ofCyclinD2,FOXE1,NPTX2,ppENK,p16andTFPI2is better than conventional MSP [45] .PanCa was predicted when methylation was observed greater than 1% in two or more of the 6 genes. Overall the panel gave 82% sensitivity and 100% specificity [45] . In order to identify biomarkers for early detection, Pedersen et al. performed a two phase study with DNA methylation signatures obtained from leukocyte DNA. The experiment was performed with 32 PDAC patients and 60 healthy controls [83] . A number of CpG sites were found to differentiate the populations. In the next phase evaluated, a panel of 5 CpG sites was shown to have potential in discriminating PDAC patients from healthy control. The sites wereIL10_P348,LCN2_P86,ZAP70_P220,AIM2_P624,andTAL1_P817.In the phase II validation study, sensitivity was 72% and specificity was 70%. The source of the samples was peripheral blood leukocyte [83] . Cai et al. found that theTNFRSF10Cpromoter was hypermethylated in all pancreatic cancer cell lines. They suggested that aberrant CGI methylation frequency in theTNFRSF10Cpromoter may be used as a biomarker of PanCa [88] . Park et al. [85] reported that CpG islands ofp16in
case of PanCa patients are methylated in 43%,ppENKin 90% andRASSF1Ain 64%. Sato et al. [89] also reported thatUCHL1,NPTX2,andSARP2were methylated in 100%, 98%, and 95% of primary PanCa tissues, respectively, ( Table 5 ).
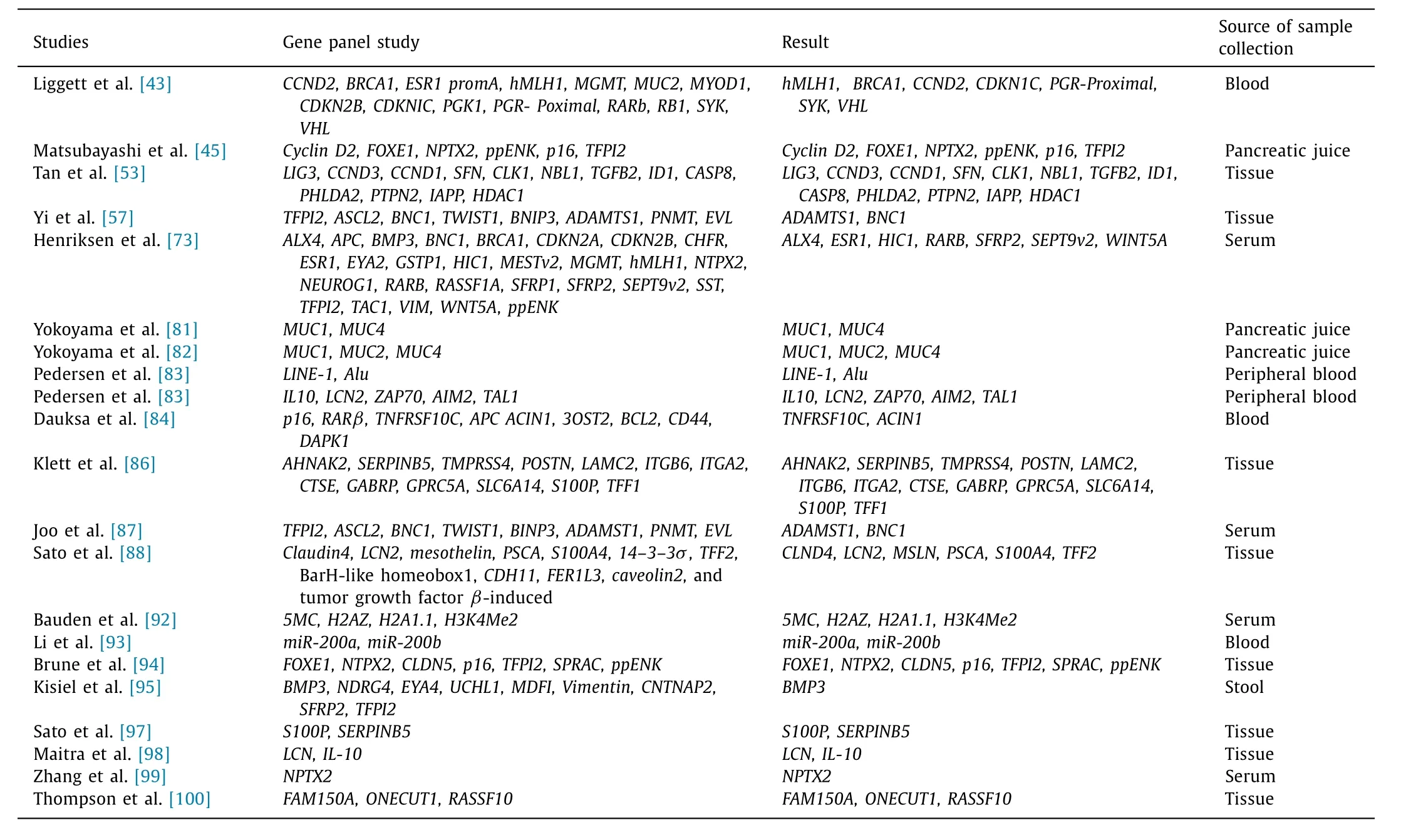
Table 5 List of epigenetic biomarkers in pancreatic ductal adenocarcinoma.
In a study evaluating correlation between multistage carcinogenesis and DNA methylation, Peng et al. has reported that overexpression ofDNMT1, a major DNA methyltransferase, is significantly correlated with accumulation of DNA methylation of tumorrelated genes and/or C-type CpG islands which are known to be methylated in a cancer-specific but not an age-dependent manner,in carcinomas derived from various organs [71] . They also observed that theBRCA1andMGMTgenes may be methylated in a tissuespecific and/or organ-specific manner in the pancreas. An explorative study by Angsuwatcharakon et al. investigated methylated DNA in the bile as a biomarker to differentiate the cause of obstruction between PanCa and benign causes [90] . For this purpose,they have collected blood from 45 patients with obstructive jaundice and divided their subject into two groups, PanCa and benign groups. They found that a methylation at cg 16,941,656 ofFRYin bile has high specificity, with an acceptable positive likelihood rate which may be helpful for distinguishing PanCa from benign tumor.According to Gao et al.,GSH2promoter methylation is highly associated with PDAC [91] .GSH2promoter was found to be hypermethylated in the PDAC tissues compared with normal pancreatic tissue from healthy subjects. Furthermore, aberrant hypermethylation ofGSH2was found to be associated with advanced stage of disease [91] . In most studies when methylation index (MI) of several genes was compared between the malignant and benign groups, they found hypermethylated genes includedDKK3,SFRP2,DKK2,NPTX2and also the large scale validation of these markers has yet to be completed. In a study of epigenetic alteration as biomarker in PDAC by Bauden et al. they reported that epigenetic alterations in the tumor genome can be detected via cfnucleosome detection panels which can be a very useful non-invasive approach [92] . Another study like epigenetic silencing of transcription factor by Li et al. 39 patients were observed with familial and 36 patients with sporadic pancreatic adenocarcinoma and pancreatic cancer cells were found to be associated with elevated expression and blood serum levels of miRs (mir200a,b) [93] . A study of Genetic and epigenetic inactivation of familiar PanCa by Brune et al.they compared the prevalence of common genetic and epigenetic alterations in sporadic and familial PDAC. On the basis of this study they reported aberrant DNA methylation of a 7 gene panel which is common in both familiar PDAC and sporadiac PDAC [94] .Liggett et al. used MethDet56 methylation analysis technique. They selected 14 gene promoters for distinguishing chronic pancreatitis from PanCa. This method yield 91.2% sensitivity and 90.8% specificity [43] . Kisiel et al. evaluated stool epigenetic alteration ofBMP3and found that the AUCs for methylatedBMP3, mutantKRAS, and combination in stool were 0.73, 0.75, and 0.85 respectively [95] .An extensive study on methylation of two different genes in pancreatobiliary fluids has been performed by Kato et al. Their result showed that hypermethylation of theUCHL1andRUNX3genes in pancreatic and biliary fluids was the most useful combined marker for PanCa (87% sensitivity and 100% specificity) [96] .
Conclusion and future prospective
PDAC is one of the lethal human cancers with a very low patient survival rate. With the progress in research along with growing evidences it is clearly indicated that significant epigenetic changes including CpG island hypermethylation and hypomethylation of genes, are characteristics of PanCa. These are promising diagnostic markers and therapeutic targets. Furthermore, it is justified that a panel of biomarkers rather than a single one will provide better diagnostics insight. Technically significant results have been observedinvitroandinvivo, whereas, clinical trials of PanCa patients show disappointing results which might be due to low level of specificity of epigenetic drugs. In order to consider epigenetic drugs as future therapies, in-depth knowledge of supporting pathways in tumor as well as normal cells are required. The methylation associated silencing of the TGS genes in PDAC is a cancer marker, affecting genes in all cellular pathways. The hypermethylated sites, among others are targeted by DNA-hypomethylating agents, thereby opening up new avenue for cancer treatment. Several studies concluded the fact that gene silencing via aberrant methylation is observed in PanCa patients, which can be dated back to the precancerous lesions/precursor neoplasms. Few other studies also stated the presence of hypermethylation of multiple genes at CpG islands of PDAC patients which dates its presence in PanINs. With progress in future research, a lot of data on the epigenetic regulation of PDAC and precancerous lesions can be expected leading to improvement of epigenetic therapies. Further studies based on use of Illumina Human Methylation 450 K Bead Array for DNA methylation can provide better insights. The recent progress in techniques like gene methylation analysis is very useful as it provides insight up to single gene level that is, it provides candidate gene approach to genome-wide scale. Some advanced techniques like MSP, array methodologies, and NGS for bisulfate treatment have become principal requisite for analysis of methylation pattern [16] . Genome-wide studies like CpG island amplification followed by CpG island microarrays along with gene expression studies in PDAC cells can provide further insights when compared with control pancreatic tissues [60] . In the near future, a prognostic signature similar to other cancers should be integrated into the PanCa staging system [99] . In this review, we have archived and descriptively reported the functions of various genes that get frequently methylated in precancerous lesions and in PDAC along side their usefulness as diagnostic/prognostic markers in upcoming epigenetic targeted therapies. We also throw light on the relationship of various gene promoter methylations with PDAC progressions.Distinctive identification of population-specific epigenetic biomarkers in genomic DNA and its correlation with therapeutic potential to further support treatment of PDAC and precancerous lesions can be a boon for future generations [100] . With respect to the above mentioned information, more input regarding NGS data on DNA methylation needs to be highlighted and is not in this review scope. In the Indian population scenario, there is significant scarcity of a model for the progression of PDAC related pattern of gene expression networks, which further represents PDAC phenotype being controlled by both of these genetic and epigenetic changes. There is lack of study of epigenetic changes in precancerous lesions and in PDAC therefore providing an open research field for further investigation.
Acknowledgments
We are thankful to Prof. Bidyut Roy, Mr. Gourab Saha, Mr. Riju Ghosh, and Mr. Sanjoy kr. Dey from Human Genetics Unit, Indian Statistical Institute, and Dr. Ankita Chatterjee from National Institute of Biomedical Genomics, Kalyani for helpful discussions and valuable inputs in the preparation of the manuscript.
CRediT authorship contribution statement
Akash Bararia:Conceptualization, Data curation, Formal analysis, Investigation, Writing - original draft.Subhankar Dey:Conceptualization, Data curation, Formal analysis, Investigation,Writing - original draft.Sumit Gulati:Investigation.Supriyo Ghatak:Investigation.Shibajyoti Ghosh:Investigation.Sudeep Banerjee:Investigation.Nilabja Sikdar:Conceptualization, Funding acquisition, Supervision, Writing - review & editing.
Funding
This study was supported by grants from Department of Biotechnology, Government of India ( RLS/BT/Re-entry/05/2012 ) and Department of Higher, Education, Science & Technology and Biotechnology, Government of West Bengal, India (BT/P/Budget/RD-37/2016).
Ethical approval
Not needed.
Competing interest
No benefits in any form have been received or will be received from a commercial party related directly or indirectly to the subject of this article.
杂志排行

Hepatobiliary & Pancreatic Diseases International的其它文章
- Transjugular intrahepatic portosystemic shunt for a patient with chylothorax in cryptogenic/metabolic cirrhosis
- Hepatobiliary&Pancreatic Diseases International
- MicroRNAs and long non-coding RNAs in liver surgery: Diagnostic and therapeutic merits
- Alpha-fetoprotein and 18 F-FDG standard uptake value predict tumor recurrence after liver transplantation for hepatocellular carcinoma with portal vein tumor thrombosis: Preliminary experience
- Translationally controlled tumor protein exerts a proinflammatory role in acute rejection after liver transplantation
- Sequential transcatheter arterial chemoembolization and portal vein embolization before right hemihepatectomy in patients with hepatocellular carcinoma