氢溴酸沃替西汀有关物质的测定方法
2020-07-06杜娟花黄美容曹阳黄顺旺曹明成
杜娟花,黄美容,曹阳,黄顺旺,曹明成
作者单位:合肥创新医药技术有限公司,安徽 合肥230088
氢溴酸沃替西汀(VorticetineHydrobromide),也称氢溴酸伏硫西汀,CAS:508233-74-7,化学名为1-[2-(2,4-甲基苯硫基)苯基]哌嗪氢溴酸盐,被认为是一种新型多模型抗抑郁药物[1],是由日本武田制药和丹麦灵北公司共同开发用于治疗重度抑郁症成人病人的药物,体外研究表明,其具有可拮抗5-HT3、5-HT7及5-HT1D受体,激活5-HT1A受体,部分激活5-HT1B受体及抑制5-HT的转运的功能[2]。是第一个具备多种药效活性的抗抑郁药物,该药物尚无国产化。
我们从2013年4月开始研究氢溴酸沃替西汀。该药物的合成由2,4-二甲基苯硫酚与邻碘溴苯反应合成2-(2,4-二甲基苯基硫基)硫醚,再与哌嗪反应制备沃替西汀,之后成氢溴酸盐,得到终产品[3-8]。制备过程中产生的杂质并经结构确证有杂质A~K。见图1。
本研究测定氢溴酸沃替西汀的有关物质为外标法、主成分自身对照法,灵敏度好、准确度高,可作为原料质量控制的评价方法。
1 仪器与试药
Shimadzu LC-20A高效液相色谱仪(岛津公司,日本);AG135电子分析天平(梅特勒托利多公司,瑞士)。

图1 氢溴酸沃替西汀制备过程中的杂质结构图
氢溴酸沃替西汀对照品(批号:20131201-J,含量:99.6%)、杂质A(批号20131101,纯度97.2%)、杂质B(批号 20131102,纯度 99.4%)、杂质C(批号20131103,纯度97.3%)、杂质D(批号20131104,纯度99.6%)、杂质E(批号20131106,纯度99.2%)、杂质F(批号13-08-0137,纯度99.2%)、杂质G(批号20131107,纯度95.0%)、杂质H(批号20131105,纯度98.8%)、杂质I(批号20131108,纯度98.5%)、杂质J(批号20131109,纯度93.3%)、杂质M(批号13-08-0138,纯度95.1%);氢溴酸沃替西汀原料药(批号20150901~20150903、20160301~20160303),以上对照品中杂质F、M为SINCO PHARMACHEM提供,其余对照品及供试品均为合肥创新医药技术有限公司合成。乙腈、三乙胺为色谱纯,水为超纯水。
2 方法与结果
2.1 溶液的制备 对照品贮备液1:取杂质B、D、E、F、G、H、I、J、M对照品适量,精密称定,加乙腈溶解并稀释,制备成每1 mL含9个杂质各25 μg的混合溶液。
对照品贮备液2:取杂质B、D、E、F、G、H、I、J、M对照品与氢溴酸沃替西汀对照品适量,精密称定,加乙腈溶解并稀释,制备成每1 mL含氢溴酸沃替西汀与9个杂质各25 μg的混合溶液。
杂质对照品溶液:精密量取1 mL杂质对照品贮备液1,置50 mL量瓶中,加乙腈稀释至刻度,摇匀,即得。
供试品溶液:精密称取氢溴酸沃替西汀原料25 mg,置50 mL量瓶中,加乙腈稀释至刻度,摇匀,即得。
0.1 %对照溶液:精密量取供试品溶液1 mL,置50 mL量瓶中,加乙腈稀释至刻度并摇匀;精密量取该稀释液1 mL,置20 mL量瓶中,加乙腈稀释至刻度并摇匀,即得。
系统适用性溶液:精密量取1 mL对照品贮备液1,置50mL量瓶中,加入经精密称定的25 mg氢溴酸沃替西汀原料,加乙腈溶解并稀释至刻度,摇匀。
2.2 色谱条件及系统适用性 色谱柱Agilent C18(4.6 mm×250 mm,5 μm);柱温40℃;检测波长226 nm;流速1.0mL/min;进样量20 μL;流动相A:0.5%三乙胺溶液(用磷酸调节pH至6.0)-乙腈(90∶10),流动相B:0.5%三乙胺溶液(用磷酸调节pH值至6.0)-乙腈(10∶90),梯度洗脱程序见表1。

表1 流动相梯度洗脱条件
称取杂质A、B、C、D、E、F、G、H、J、M、I适量,制备各杂质的单标溶液,进行定位,再进注系统适用性溶液进行液相色谱分析。见图2。
系统适用性溶液中各已知杂质均能与主成分达到较好分离,分离度均良好。
2.3 专属性 取氢溴酸沃替西汀原料25 mg,置50 mL量瓶中,分别进行高温、光照、氧化、酸、碱破坏,酸、碱破坏进行中和,再用乙腈溶解并稀释定容,摇匀;并同法制备破坏空白。按“2.2”项下色谱条件进样分析。结果表明,本品在强光及高温条件下有少量降解杂质,强酸、强碱、强氧化时能产生明显降解杂质。且主成分峰与相邻杂质峰分离度能符合要求,破坏前后物料基本守恒,主峰纯度符合要求。

图2 系统适用性试验色谱图
2.4 定量限、检测限、线性范围与校正因子 精密量取对照品贮备液2,加乙腈稀释成0.2~2.0 μg/mL不同浓度的系列对照品混合溶液,分别进样,记录色谱图。以各成分的峰面积为纵坐标(Y),浓度为横坐标(X),进行线性回归,并以回归方程的斜率计算各已知杂质相对于氢溴酸沃替西汀的校正因子(RCF),同时测定各成分的定量限(LOQ)与检测限(LOD),结果见表2。
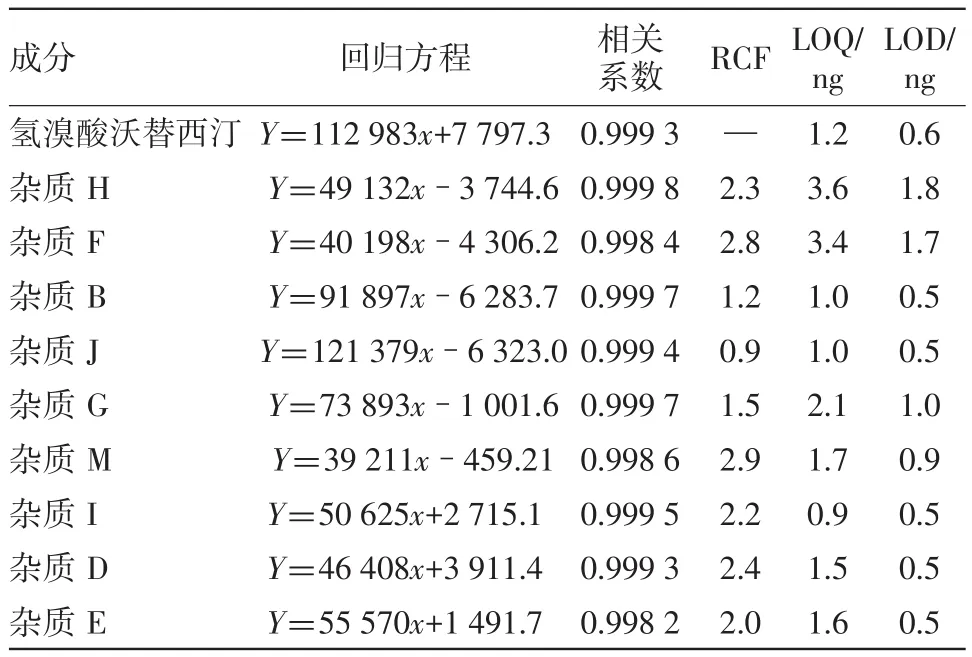
表2 氢溴酸沃替西汀有关物质线性和RCF
2.5 精密度与准确度 精密度:按“2.1”项下方法由双人各制备供试品6份,在“2.2”色谱条件下,以不同液相色谱仪进行测定,计算各杂质RSD。单人、6份样品,总杂含量的RSD均<7.5%,单个杂质含量的RSD均<10%。双人共12份样品,总杂含量的RSD均<10%,单个杂质含量RSD均<15%。
精密量取杂质对照品贮备液1,加入供试品中,制成含各已知杂质为限度的50%、100%、150%的回收率样品,进样分析,记录色谱图,计算各杂质回收率。9个已知杂质B、D、E、F、G、H、I、J、M的平均加样回收率在(100.2±1.1)%。
2.6 溶液稳定性 按“2.1”项下制备杂质对照品溶液与供试品溶液,25 ℃放置,并于0、2、4、8、12、24 h分别取样进样分析,记录色谱图。计算得杂质对照品溶液中单个杂质峰面积的RSD均<4%,供试品溶液中,各杂质峰面积的RSD均<15%,且无新杂质产生。
2.7 耐用性 通过分别调整色谱条件中的柱温±2℃、检测波长±2 nm及pH值±0.2,考察系统适用性溶液中杂质与主成分之间分离度的情况。结果表明,柱温、流速及pH值等参数的调整对测定结果基本不会产生影响,方法的耐用性良好。
2.8 样品测定 按上述已确定的色谱条件及方法,对氢溴酸沃替西汀原料6批进行有关物质测定,按外标法计算供试品溶液中已知杂质含量,自身对照法计算未知杂质含量。其中,杂质B~I均未检出。其他杂质检测见表3。
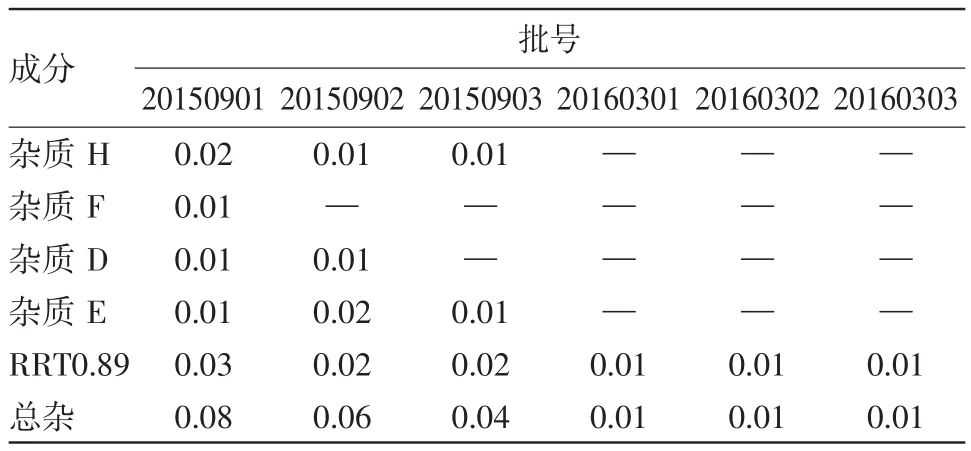
表3 氢溴酸沃替西汀有关物质检测结果/%
3 讨论
以乙腈为溶剂,分别配制氢溴酸沃替西汀和9个已知杂质(杂质B、D、E、F、G、H、I、J、M)的对照品溶液,在190~400 nm波长范围内进行光谱扫描,氢溴酸沃替西汀和杂质B、G、J)在226 nm处有最大吸收,杂质D、E、F、H、I、M在226 nm处均有较大吸收,故采用226 nm作为检测波长。根据专利[9]中有关物质,流动相为甲醇:乙酸铵缓冲液的反相体系;为了改善各杂质的峰形,将流动相中缓冲系统定为磷酸-三乙胺缓冲体系,进行优化后确定梯度洗脱程序,各组分的峰形良好,分离度符合要求,故选择该流动相体系。
国外药典尚未收载氢溴酸沃替西汀原料或制剂标准,在已有文献[10-14]报道的有关物质研究中,有HPLC法、液质联用法、ESI-Q-TOF/MS、APCI-TOF/MS等方法,检测的杂质数量2~7个,涉及的杂质含本研究中的杂质B、E、F、I、M。本研究中由于杂质A与杂质C分别是由起始原料2,4-二甲基苯硫酚中的2,5-二甲基苯硫酚与3,4-二甲基苯硫酚参与化学反应产生的副产物,且单标定位时杂质A、C与主峰的出峰位置相同,故可通过控制起始原料2,4-二甲基苯硫酚的产品质量来控制成品中杂质A、杂质C的含量;杂质G、J为邻溴碘苯中杂质间溴碘苯、对溴碘苯分别参与反应生成的,与氢溴酸沃替西汀为苯环上位置异构杂质,分离难度较大;杂质D、H为邻溴碘苯与哌嗪进行反应产生的杂质。本研究在液相色谱条件下氢溴酸沃替西汀、杂质G、杂质J依次出峰,与相邻峰均能达到很好地分离。
已知杂质B、D、E、F、G、H、I、M的校正因子不在0.9~1.1范围内,参照文献[15],建议将已知杂质订入质量标准,按外标法计算、质控限度为0.1%;未知杂质按自身对照法、0.1%的限度进行控制。
