供电子基团修饰对NNI-R系列分子光物理性质的影响
2020-07-05林桂锋郭景富何腾飞任爱民
林桂锋, 郭景富, 何腾飞, 任爱民
(1. 东北师范大学物理学院, 长春 130024;2. 吉林大学理论化学研究所, 长春 130061)
磷光材料在OLED、 传感、 生物成像以及DNA检测等显示和信息处理方面具有重要应用[1~5]. 1998年, Forrest等[6]将八乙基卟啉铂(PtOEP)作为发光层掺杂在主体材料八羟基喹啉铝(Alq3)中, 得到了内、 外量子效率分别为23%和4%的饱和红色磷光器件[6]. 这种混合了单重态和三重态激子的磷光发射, 其量子效率突破了传统有机电致荧光材料低于25%的限制, 理论上允许内量子效率高达100%. 从此金属络合物发光材料以潜在的高发光效率的特点受到广泛关注[7~11]. 在有机电致磷光材料的研究中, 除过渡金属络合物, 卤素原子以其固有的重原子效应也受到广泛关注[12]. 这主要与磷光发射的机制有关, 已知单重激发态到三重激发态的跃迁是禁阻的, 所以提高三重态产率、 加强磷光的有效方法就是增强系间窜越的效率[13,14]. 目前, 通常通过添加过渡金属原子或卤素重原子的方法来增强系间窜越, 有机分子中较重的顺磁性金属离子或卤素原子可加强旋轨耦合作用, 从而促进系间窜越[15~18]; 金属体系的跃迁有金属d轨道的参与, 这种金属到配体或配体到金属的电子转移激发态有利于从单重态到三重态的跃迁[18,19].
近年来, 铂、 铱和钯等有机金属体系的磷光发射材料有了很大发展[20~27], 但其固有的缺陷, 如重金属原子的毒性[28~30], 过渡金属-碳键的不稳定性[31], 以及贵金属成本过高等问题限制了这些金属分子的应用. 相比之下, 纯有机分子由于其毒性小, 成本低以及可加工灵活性高已成为一种替代选择. 由于振动失活和氧气猝灭等因素, 纯有机分子的三重态激子很容易发生非辐射衰减, 所以纯有机磷光材料的研究还仅限于惰性和冷冻低温环境中. 因此, 研究新型纯有机室温磷光材料成为有机电致发光材料的热点. 1,8-萘酰亚胺(1,8-Naphthalimides, NIs)具有非常有趣的光物理性质以及广泛的生物学适用性, NIs系列的分子被广泛用作蓝光二极管材料[32~34]. 当在NIs中引入不同的吸电子(或供电子)基团时, 会显著改变分子激发态给体和受体之间的二面角, 进而改变光致发光波长和量子产率等光物理性质[33~37]. 因此, NIs分子作为双荧光发射分子被大量研究[35~38]. 2004年, Berces等[38]在三乙酸甘油酯溶液中, 测定了甲氧基对N-苯基-2,3-萘酰亚胺的苯基对位氢原子进行取代后化合物的发光光谱, 发现有明显的双荧光发射现象, 并将长波荧光发射部分归结为分子内电荷转移(ICT)的结果[38]. Zhang等[39]在羰基化合物修饰的二氟硼-β-二酮配合物上观测到荧光-磷光双发射现象, 其激发态同时具有LE和ICT 2种电子激发态性质. 他们利用不同供电子能力的基团对1,8-Naphthalimide进行修饰, 成功合成了一系列室温磷光材料[40]. 在N-苯基-1,8萘二甲酰亚胺(N-Phenyl-1,8-Naphthalimide, NNI-ph)上用甲氧基对苯基对位氢原子以及对位和间位氢原子进行取代, 合成了MeOPh和MeO3Ph分子, 并将其掺杂到聚甲基丙烯酸甲酯(PMMA)制成的固态薄膜中, 成功测得磷光发射的特异现象. 基于此, 他们提出了一种新的纯有机室温发光材料的合成方案: NNI可以通过给体修饰引入ICT激发态, ICT态的HOMO和LUMO重叠性较差, 可以进一步缩小单线态和三线态之间的能差, 从而促进系间窜越速率, 产生室温磷光[40].
Barbatti等[41]用吸电子和供电子基团分别对NNI-ph的苯基对位和间位氢原子进行取代, 在二氯甲烷溶剂中, 采用连续极化介质模型(PCM), 用密度泛函理论(DFT)和含时密度泛函理论(TD-DFT)方法对2种不同性质基团修饰分子的荧光速率(kFL)和系间窜越速率(kISC)进行理论计算, 结果表明, 供电子基团修饰分子引入的ICT激发态可以大幅抑制荧光发射, 并促进单三重态系间窜越的发生, 为供电子基团取代分子的室温磷光现象提供了合理解释.
但是, Barbatti等[41]在关于NNI-ph苯基对位氢原子的取代研究中, 仅选取2个供电子基团(羟基和甲氧基), 他们的结论普适性有待验证, 为此, 本文以NNI-ph为基础设计了一系列的供电子基团修饰的NNI-ph-R分子(NNI-R), 并分别在极性溶剂和气相环境中, 通过量子化学计算从理论上验证这种设计方法的合理性以及普适性.
1 计算模型及方法
图1给出了以NNI-ph(NNI-H)为原型分子, 用不同供电子基团(甲基、 叔丁基、 羟基、 甲氧基、 胺基和二甲基胺基)对苯基对位氢原子进行取代, 并分别命名为NNI-Me, NNI-tBu, NNI-OH, NNI-OMe, NNI-NH2和NNI-NMe2, 分析不同基团的供电子能力与NNI-R系列衍生物分子的结构变化、 电子跃迁性质以及荧光速率和系间窜越速率的关系, 进而研究供电子基团取代对分子的电子跃迁性质及发光性质的影响.
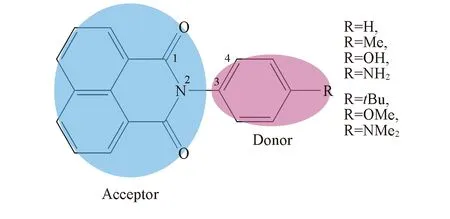
Fig.1 Chemical structures of NNI-R
采用PCM溶剂模型[41,42], 计算了二氯甲烷溶剂中NNI-OH, NNI-OMe和NNI-NH2分子的最低单重激发态(S1)的几何结构以及电子结构. 计算采用Gaussian 程序[43]的TD-DFT方法完成[44]. 结构优化及能量计算均采用ωB97XD泛函[45], 几何优化采用6-31G*基组, 能量计算采用6-311+G**基组[46,47].
考虑到溶剂环境下电荷和电荷重组过程的速率大小主要取决于溶剂的极性[48], 与实验光谱测定的薄膜环境差异较大, 为摒除溶剂极化环境的影响, 在气相环境下对NNI-R系列全部分子进行理论研究. 全部分子的电子结构均采用Gaussian程序[49]的DFT和TD-DFT方法完成[44], 并根据实验重新评估了泛函的适用性(泛函选择见表S1, 见本文支持信息), 结果表明, 具有54%HF成分的M06-2X泛函在局域激发和电荷转移激发单重态的计算以及三重态的计算方面均具有较强的适用性. 因此, 基态和激发态几何优化和能量计算均采用了M06-2X泛函[50~52]. 由于几何优化对基组不敏感, 采用小基组便于节省计算时间, 所以几何优化采用6-31G*基组, 能量计算采用6-311+G**基组[46,47]. 考虑到研究体系涉及到电荷转移的电子激发特征, 为准确描述激发态能量变化, 在结构优化和能量计算过程中均引入零阻尼色散校正D3[53,54], 所有分子的基态和激发态优化结构的频率均为正频, 确保分子结构的稳定性.
采用Multiwfn程序[52]将TD-DFT提供的非驰豫激发态组态系数进行可视化来表现电子和空穴密度分布[55]. 分子的旋轨耦合矩阵利用PySOC程序进行计算[56].
本文旨在探讨在单个分子水平上不同发光类型的基本机制, 研究不同基团取代时, NNI-R系列分子的荧光速率和系间窜越速率的变化. 不涉及分子间相互作用和堆积效应, 以及固态薄膜对单分子外在压力的影响.
2 结果与讨论
2.1 极性溶剂中NNI-OH, NNI-OMe和NNI-NH2分子的理论计算
采用与文献[41]相同的计算方法对NNI-OH和NNI-OMe分子进行优化, 同时计算了强供电子基团(胺基)取代分子S1态的分子结构和能量(表1)以及电子跃迁性质(图S1, 见本文支持信息). 表1列出了给体和受体之间二面角(C1—N2—C3—C4)的大小和分子的总能量.
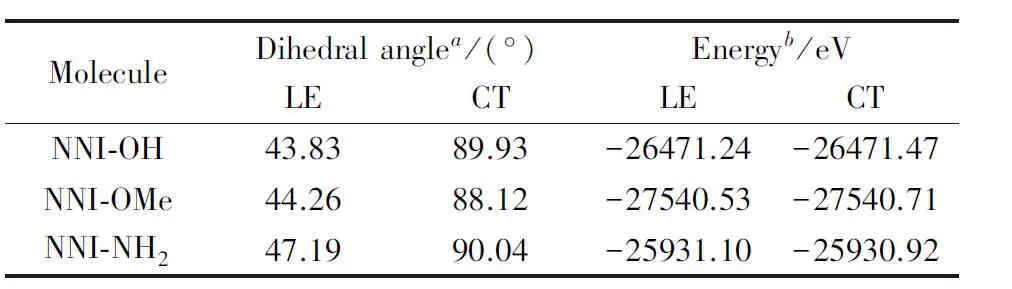
Table 1 Dihedral angle and energy atoptimized S1 of NNI-R
根据优化结果可知, 在苯基对位氢原子取代时, 羟基、 甲氧基和胺基取代分子的S1态均存在异构体, 一种为给体和受体之间二面角为44°~48°的近平面结构, 表现为电荷转移激发态(CT); 另一种为二面角约90°的垂直结构, 表现为局域激发态(LE)(图S1). 文献[41]中仅给出给体和受体之间二面角约44°的结构, 由表1可知, 羟基和甲氧基取代分子的S1态的LE结构的能量远低于CT结构, 分别低0.23和0.18 eV. 相反, 胺基取代分子的S1态的2个异构体中, LE结构的能量比CT高0.18 eV.
根据热力学结果分析, 在溶剂极化下萘二甲酰亚胺给体取代物中不是所有CT结构都最稳定, 与在PMMA中的实验现象有可能不一致, 因此有必要进行补充和完善, 以澄清实验现象本质. 可能主要与二氯甲烷溶剂的强极化性质有关(实验现象在PMMA薄膜中测定), 溶剂的极化作用不利于给体的扭转, 在弱供电子基团(羟基、 甲氧基)取代时, 分子S1态的给体和受体之间二面角垂直, 分子的激发态会呈现局域激发, 不利于系间窜越. 因此, 在气相环境下对NNI-R系列分子进行了理论研究.
2.2 气相环境中NNI-R系列分子的理论计算
2.2.1 分子的基态和激发态结构以及前线分子轨道分布 对于单键联结的D-A结构有机小分子, 激发态电子跃迁性质与给体和受体之间二面角的大小密切相关, 二面角若近垂直, 一般呈局域激发; 若二面角角度小、 近共平面结构, 则呈现电荷转移激发[35,37,38,57]. 由于几何结构和电子结构对单重态和三重态之间的能隙以及相关的ISC过程具有很大影响, 在NNI-R系列分子的研究中将供电子基团和苯基片段作为给体部分(D), 萘二甲酰亚胺本体作为受体(A)(图1), 分别对基态(S0)和S1态的几何结构和电子结构进行分析. 将S0态和S1态下给体和受体之间的N2—C3键长和C1—N2—C3—C4二面角以及其变化列于表2.
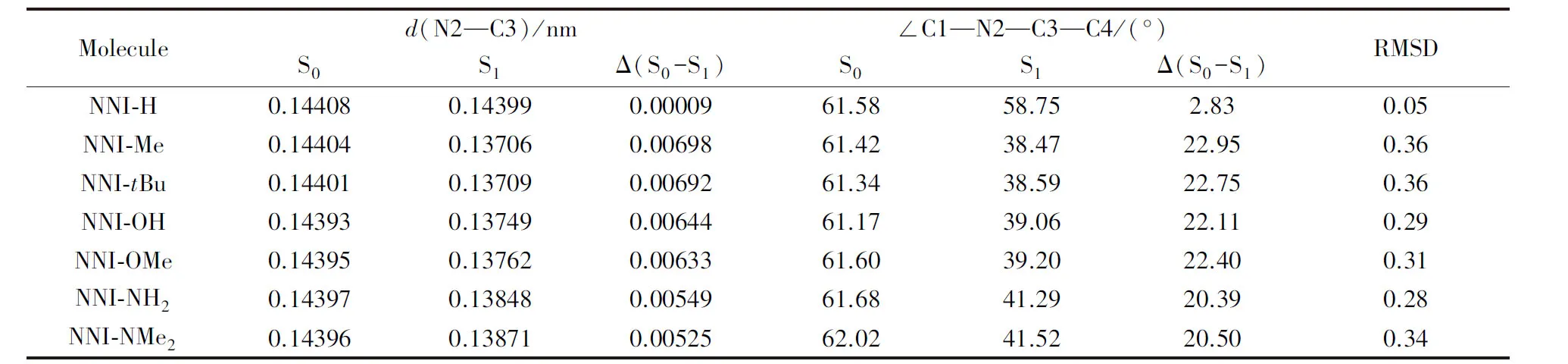
Table 2 Bond length and dihedral angle between the donor and the acceptor, and the root meansquare deviation(RMSD) at optimized S0 and S1 of NNI-R
由表2可见, NNI-H分子激发态和基态构型无显著差异, 而当对苯基对位氢原子进行供电子基团取代后, S1态给体和受体之间的二面角从61°减小至38°~42°, 减少了约20°, 且呈现为CT激发, 这种二面角向平面化转变趋势引起的电子跃迁性质的改变被称为平面化的分子内电荷转移(PLICT). 这种供电子基团取代的分子的S1态经历了从基态到激发态的剧烈结构变形, 从NNI-Me至NNI-NMe2的S1态和S0态的较大结构差异[Δ(S0-S1)和均方根偏差(RMSD)值均较大]可能会抑制分子S1→S0的荧光发射. 不同激发态结构变化会导致分子发光性质的改变.
由图2的S0态前线分子轨道分布可见, 在含有供电子基团的NNI-Me至NNI-NMe2分子中, 最高占据轨道(HOMO)和最低未占据轨道(LUMO)电子云重叠度很低, HOMO与LUMO明显分离, 分子的电子耦合作用明显变弱. 供电子基团取代对能级的影响主要体现在HOMO能级上, 取代基供电子能力越强, 激发态HOMO能级越升高, 引起能隙减小; 随着HOMO-LUMO能隙逐渐减小, 系间窜越的能垒即单-三重激发态能差ΔES1-T1和ΔES1-T2(表3)均逐渐减小, 极大地增强了发生系间窜越的可能.
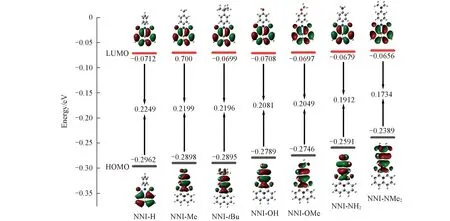
Fig.2 Plots of frontier molecular orbitals, together with the HOMO, LUMO energylevels and their gaps at optimized S0 of NNI-R
Table3Calculatedadiabaticenergydifference(ΔEST)betweenS1andT1/T2stateofNNI-R

2.2.2 激发态电子跃迁性质和旋轨耦合常数计算 为了直观地分析S1态的电子跃迁特性, 在图3中用Multiwfn程序描绘了S1态的空穴和电子分布, 蓝色表示空穴, 绿色表示电子.
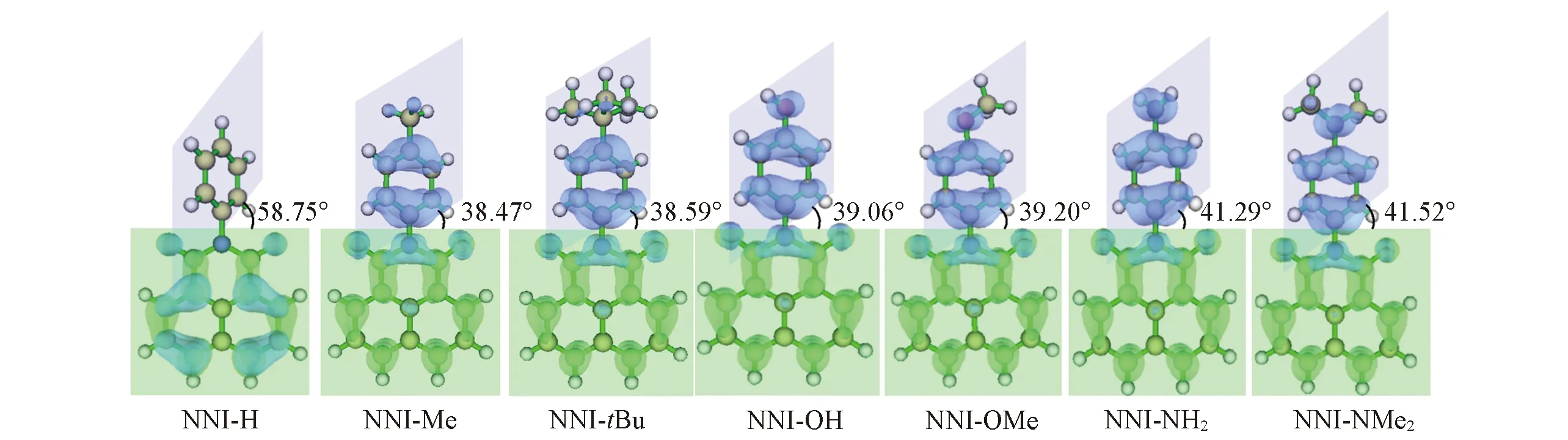
Fig.3 Hole-electron distributions of NNI-Rs atoptimized S1 Blue: holes; green: electrons.
结合表4和图3的分子空穴和电子的密度分布可见, NNI-H的电子跃迁参数与其它衍生物显著不同, 其空穴和电子质心之间距离的D指数小,Sr较大,t指数明显为负, Δσ指数不大. 这种激发的空穴-电子主要分布范围很接近, 重叠较高(Sm和Sr较大), 空间和电子分布没有明显分离, 因此NNI-H的S1态是典型的局域激发. 而当对苯基氢原子进行供电子基团取代时,D指数较大,Sr较小,t指数为正, Δσ指数较大, 表明它们的S1态是单向的CT激发. 取代基不同影响了空穴和电子的重叠程度, 相比较而言, NNI-NH2, NNI-NMe2的空穴和电子空间重叠程度最小, 分布广度最小(Δσ指数小), 而分离度最大(t指数), 并且空穴和电子质心之间距离最大(D指数大). 其次为NNI-OH和NNI-OMe, 最后为NNI-Me和NNI-tBu. 即从空穴和电子的整体空间分布来看, 从NNI-Me到NNI-NMe2, S1态的CT激发性质逐渐明显化(Sm和Sr减小,t指数为正且增大,D指数增大).

Table 4 Main parameters about the hole-electron distributions of NNI-R*
* All parameters are calculated by Multiwfn.a.Sm,index: The overlap function of hole and electron distribution is integrated in full space;b.Sr,index: the geometric average of overlap function of hole and electron distribution is integrated in full space;c.tindex: the separation distance of hole and electron in CT direction;d.Dindex: the distance between the centers of mass of hole and electron;e. Δσindex: the difference of spatial distribution extent between hole densities and electron densities in full space.
从图3可知, 所有供电子基团取代分子的S1态均表现为ICT激发, 电荷在萘二酰亚胺本体和给体(供电子基团加苯基片段)之间转移, 表5列出了NNI-H至NNI-NMe2分子在S0态和S1态下供电子基团和给体的电荷密度分布, 分析NNI-R系列分子的S1态相对S0态的电荷转移发现, 在NNI-H中, 苯
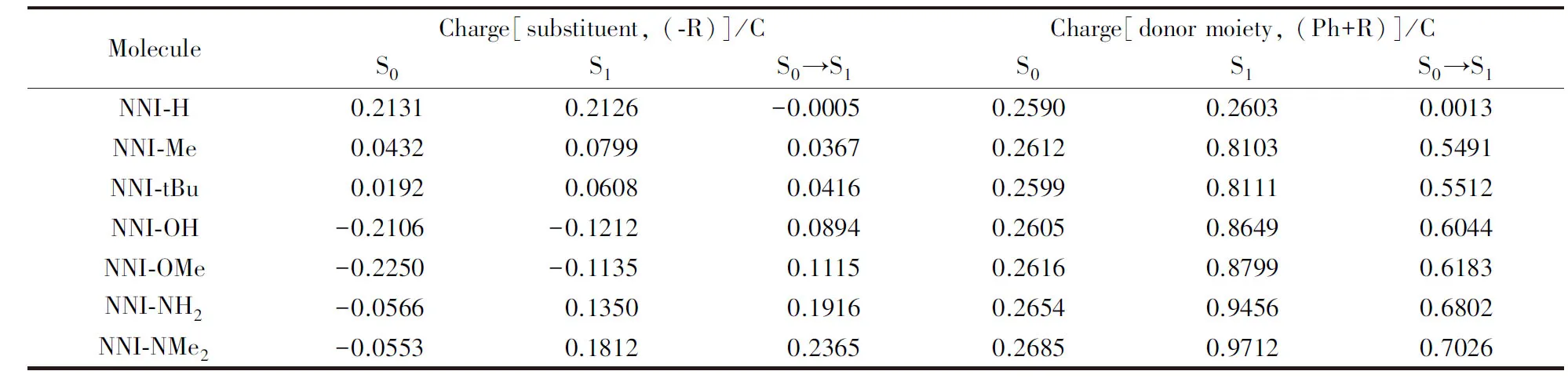
Table 5 Natural charge distribution of the substituents and the donor moieties atoptimized S0 andS1 state, and charge transfer from S0 to S1 state of NNI-R
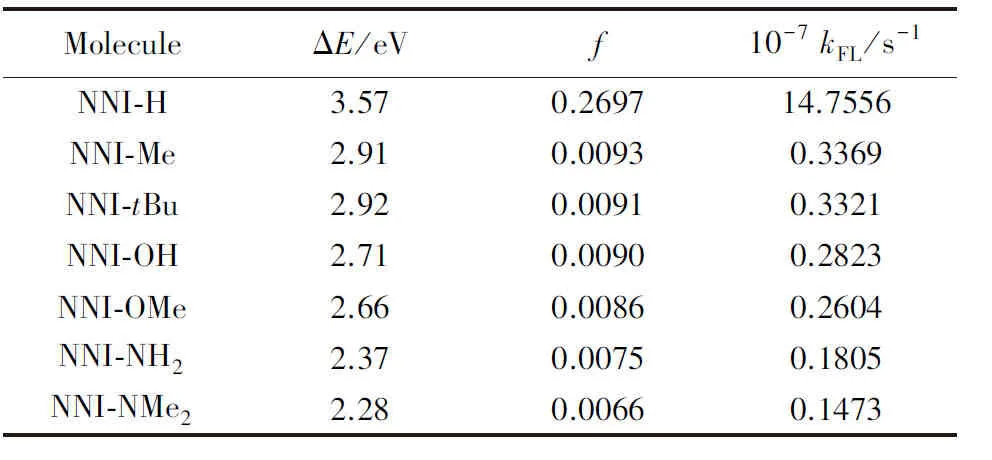
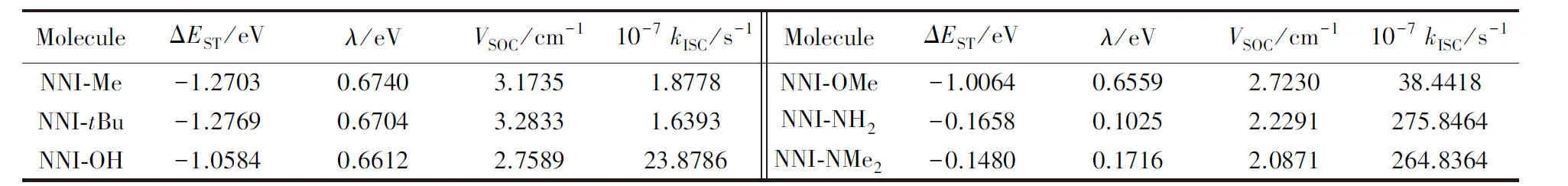
基对位氢原子电荷转移量近似为零, 即没有供电子能力, 而在NNI-Me至NNI-NMe2中, 供电子片段上电荷转移逐渐增大, 取代基团的供电子能力顺序为Me 图4给出了供电子基团取代分子的取代基上的电荷变化量、 给体上的电荷变化量以及D指数的变化. 可见, 随着取代基团供电子能力增强, 给体的电荷转移量逐渐增大, 电子和空穴的整体空间分布越广, 在CT方向上空穴和电子分离程度增大(Sm和Sr减小,d指数为正且增大,D指数增大), 分子的S1态电荷转移激发性质变强. 综上, 不同分子的电荷转移强度顺序为NNI-H< Fig.4 Charge change of substitutents and donor moieties during from S0 to S1 state and the change of Dindexa. NNI-Me; b. NNI-tBu; c. NNI-OH; d. NNI-OMe; e. NNI-NH2; f. NNI-NMe2. 2.2.3 荧光速率和系间窜越速率计算 用量子化学方法计算分子的荧光速率(kFL)和系间窜越速率(kISC)是研究室温有机发光材料的重要手段. 在典型的有机分子中, 初始态和最终态的自旋-轨道相互作用与其绝热能差相比相对较小, 并且在初始态为激发态时, 高振动能级的密度较高. 因此, 可以采用费米黄金规则结合微扰理论推导出的ISC(最低单线态和三线态)速率公式来计算. 在电子转移和能量转移中, 跃迁机制都涉及驱动反应物结构变化的振动运动, 在Franck-Condon近似下将速率表达式分解为电子和振动部分[55]: 式中:λ为由分子振动引起的内重组能和溶剂引起的重组能(未考虑溶剂);kB(1.38×10-23J/K)为玻尔兹曼常数; ΔEST(eV)为单重态和三重态的能量差;T(室温约为300 K)为温度. 荧光速率(kFL)用爱因斯坦公式表示为[41] 式中:ε0(8.85×10-12F/m)为真空介电常数;me(9.11×10-31kg)为电子质量;c(3.0×108m/s)为光速;e(1.60×10-19C)为元电荷;f为S1态发射的振子强度; ΔE(eV)为S1态的激发能. Table6Calculatedfluorescentrate(kFL),withtheexcitationenergy(ΔE)andoscillatorstrength(f)ofS1stateofNNI-R 利用爱因斯坦公式计算分子的荧光速率列于表6, 当苯基对位氢原子被供电子基团取代时, S1态的振子强度从0.2697陡降至0.0066. 比较NNI-Me到NNI-NMe2, 给体部分CT态的性质越明显, S1→S0跃迁偶极矩变小, S1态激发能也减小. 在供电子基团取代氢原子后, 分子的荧光速率下降两个数量级, 表现为荧光发射剧烈降低, 显示出非常弱的荧光现象. 给体的电荷转移能力与荧光发射速率大小密切相关. 由表6可以看到, 从NNI-Me到NNI-NMe2, 随着取代基给电子能力的逐渐增强, 电荷在CT方向上移动距离增大(表4中d指数增大), S1→S0荧光过程的CT激发性质逐渐增强(f逐渐减小), 电子-振动耦合作用增强(λ增大), 抑制荧光发射的能力也随之增强. 给体的电荷转移能力与分子的系间窜越速率有着密不可分的关系. 随着给电子能力的逐渐增强, S1→T1/T2的系间窜越几率有可能大大增强. 对于不含重原子的有机小分子体系, 在系间窜越过程中, 由于电子自旋翻转而导致自旋角动量发生改变, 为了保持体系总角动量守衡, 必然要发生单中心效应, 即单中心的px→py轨道间跳跃. 具有单中心效应的体系能够优先保证系间窜越的发生. 因此, 根据El-Sayed规则, 单重态和三重态应由不同的轨道跃迁类型组成, 系间窜越需遵循以下选择规则: 对S1→ T1的系间窜越:1(n,π*)→3(π,π*), 允许;1(π,π*)→3(n,π*), 允许;1(n,π*)→3(n,π*), 禁阻;1(π,π*)→3(π,π*), 禁阻. 对T1→S0的系间窜越:3(π,π*)→n2, 允许;3(π,π*)→π2, 禁阻. 计算S1态到T1态和T2态的旋轨耦合矩阵元列于表7, 可见, 对于NNI-Me到NNI-OMe, 最大的单三重态旋轨耦合作用发生在S1-T1之间. 而对于NNI-NH2和NNI-NMe2, 最大的旋轨耦合作用发生在S1-T2态之间, 这种反常的系间窜越现象主要是由El-Sayed跃迁规则决定. 已知萘二酰亚胺上的N原子的未成键孤对电子有利于系间窜跃, 根据图5自然轨道跃迁(NTO), 分析了N原子上占据轨道(空穴)的密度分布, 从而讨论激发态跃迁类型, 如处于S1态时, NNI-H分子的N原子上无占据轨道分布, Table 7 Calculated spin-orbit coupling(SOC) matrix elements(cm-1) at optimized S1 geometry Fig.5 Hole-electron of NNI-R in S1, T1 and T2 state at optimized S1 state 推测NNI-H分子的S1态为1(π,π*), 而处于S1态的NNI-Me分子的N原子上存在占据轨道分布, 且方向与苯基π电子云垂直, 即NNI-Me分子的S1态为3(n,π*). 根据上述方法, NNI-H分子的T1态和T2态均为3(π,π*), 因此NNI-H分子的单重态和三重态之间的系间窜越是禁阻的. 对于NNI-NH2和NNI-NMe2, S1态的跃迁表现为1(n,π*), T1态为3(n,π*), T2态为3(π,π*), S1→ T1禁阻, S1→ T2允许. 相反, NNI-OH, NNI-OMe, NNI-Me和NNI-tBu, S1态的跃迁为1(n,π*), T1态为3(π,π*), T2态为3(n,π*), 因此S1→ T1允许, S1→ T2禁阻, El-Sayed跃迁规则下的S1态和T1/T2态之间的系间窜越与旋轨耦合常数的大小相对应. 将S1态作为初始态, T1/T2态作为末态, 分别计算了S1-T1和S1-T2的绝热能差(表3). 分子NNI-NH2和NNI-NMe2的T2态电子能级更接近S1态, 较低的能级差有效促进了S1态到T2态的系间窜越. 由表7可以确定旋轨耦合作用最强的态, 利用半经验的Marcus公式分别计算NNI-Me至NNI-OMe的S1→T1的系间窜越速率以及NNI-NH2和NNI-NMe2的S1→T2的系间窜越速率. 由表8可见, 在胺基和二甲胺基取代时, 系间窜越速率显著增大, 主要是由于单三重态绝热能差降低和重组能减小导致. 从激发态结构上看, 胺基和二甲胺基的S1态和T2态的给体和受体之间二面角变化较小(表S2, 见本文支持信息), 振动弛豫不明显, 是导致重组能降低的重要因素. Table8CalculatedISCrate(kISC)withthesinglet-tripletenergygap(ΔEST),thereorganizationenergy(λ),andthespin-orbitcouplingmatrixelement(VSOC)* * All calculations are carried out between S1and T1state for the first four molecules and between S1and T2state for the last two molecules. 对比表6中分子的荧光速率, NNI-Me至NNI-NMe2分子的S1→T1/T2激发态的ISC速率比荧光速率增大1~3个数量级. 由于竞争机制的存在, 供电子基团取代时显著增大了ISC速率, 使单重态到三重态的系间窜越成为一条更可能的跃迁路径, 为NNI-R供电子基团取代分子的室温磷光发射提供了合理解释. 采用DFT和TD-DFT方法研究了一系列供电子基团取代修饰的NNI-R分子在气相条件下的发光行为, NNI-H表现为荧光发射, 是萘二酰亚胺本体上局域激发的结果. 供电子基团取代时, NNI-R分子的S1态具有的电荷转移激发性质导致荧光猝灭. 同时, 取代基供电子能力越强, 给体的电荷转移越多, CT方向上空穴和电子的距离越大, 分子内电荷转移激发强度越大, 抑制荧光发射的能力越强. 在供电子基团取代时, 分子的系间窜越速率远大于荧光速率, 这使NNI-R系列分子的磷光发射成为一种可能. 与实验现象完全一致, 即在气相或固态PMMA薄膜中, 用供电子基团修饰NNI-R分子可以有效促进系间窜越. 研究结果可为基于NNI的纯有机室温磷光材料的合成和表征提供重要的理论依据, 但该结论不适用于极性溶剂条件. 因为溶剂的极化作用不利于给体的扭转, 在极化溶剂中, 弱供电子基团(—OH和—OMe)修饰的NNI-R分子的S1激发态给体和受体间之间二面角相互垂直, 分子的激发态会呈现局域激发态, 从而抑制了系间窜越的出现, 因此不会发生室温磷光现象. 支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20200086.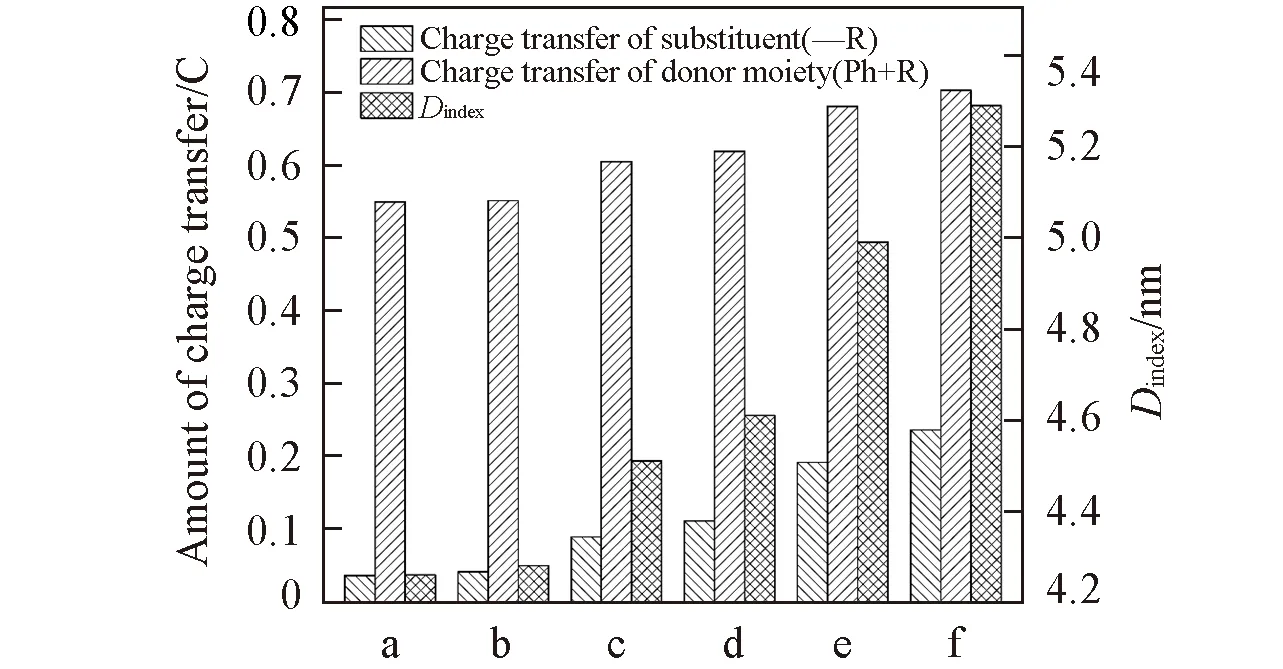
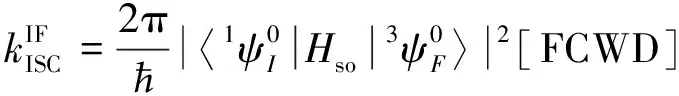
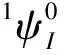

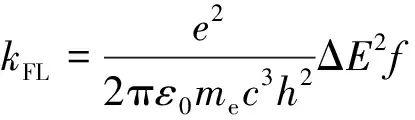


3 结 论
