基于网络药理学探讨降压通脉方的作用机制
2020-06-24张帆韩琳宋林梅李冰沈一凡郭维琴秦建国
张帆 韩琳 宋林梅 李冰 沈一凡 郭维琴 秦建国


摘要 目的:运用网络药理学理念和方法探讨名中医效方降压通脉方的可能靶点及作用机制,加深对名中医效方的理解。方法:通过检索ETCM及BATMAN-TCM数据库获取降压通脉方成分及预测靶基因;采用STRING、Cytoscape软件及插件构建网络并分析。利用DAVID工具对关键靶点进行基因本体论(GO)功能富集分析、KEGG通路富集分析以及疾病(DO)富集分析。结果:从降压通脉方中筛选出关键化合物112个,Hub靶标113个,模块29个。主要涉及在炎性反应、信号转导、免疫、糖脂代谢等生物学过程中的多条信号通路。结论:初步揭示了降压通脉方主要活性成分、靶点及作用机制,为进一步深入探讨其药理学作用提供了参考。
关键词 降压通脉方;名医经验;网络药理学;靶点;信号通路;生物功能
Study on the Action Mechanism of Jiangya Tongmai Formula Based on Network Pharmacology
ZHANG Fan1,HAN Lin2,SONG Linmei3,LI Bing1,SHEN Yifan1,GUO Weiqin4,QIN Jianguo5
(1 Graduate School,Beijing University of Chinese Medicine,Beijing 100029,China; 2 College of Traditional Chinese Medicine,Beijing University of Chinese Medicine,Beijing 102488,China; 3 Capital Medical University Yanjing Medical College,Beijing 101300,China; 4 Dongzhimen Hospital,Beijing University of Chinese Medicine,Beijing 100700,China; 5 Nephrology Department,Dongfang Hospital,Beijing University of Chinese Medicine,Beijing 100078,China)
Abstract Objective:To investigate the possible action mechanism of Jiangya Tongmai Formula based on network pharmacology.Methods:By searching the ETCM and BATMAN-TCM databases,the components of Jiangya Tongmai Formula and the predicted target genes were obtained; the network was constructed and analyzed by STRING,Cytoscape software and plug-ins.DAVID tools was used to perform gene ontology(GO)function enrichment analysis,KEGG pathway enrichment analysis,and disease(DO)enrichment analysis on key targets.Results:From Jiangya Tongmai Formula,112 key compounds,113 Hub targets and 29 modules were screened.It mainly involved multiple signal pathways in the biological processes of inflammation,signal transduction,immunity,and glucose and lipid metabolism,and many other signal path.Conclusion:The main active ingredients,targets and mechanism of Jiangya Tongmai Formula are initially revealed,which provides a reference for further in-depth study of its pharmacological effects.
Keywords Jiangya Tongmai Decoction;Experience of famous doctor;Network pharmacology;Targets;Signaling pathway;Biological functions
中圖分类号:R285文献标识码:Adoi:10.3969/j.issn.1673-7202.2020.23.014
降压通脉方是著名中医药学家郭士魁先生创制的效方[1],主要用于治疗冠心病合并高血压病的患者。该方以丹参为君药,配伍红花、郁金及鸡血藤凉血活血,行气通络止痛,合用珍珠母平肝潜阳,黄芩、菊花等清肝泻火,诸药合用共奏活血祛瘀、平肝潜阳之功[1-2]。异病同治是中医治疗的一大特点,基于对疾病证候的认识,诸多医家将其应用于治疗高血压心脏病、高血压肾损害等病症的治疗,取得了良好的临床疗效。目前已基于此进行了一系列的临床及实验研究[2-12],证实该方能增强左室舒张功能、左室收缩功能、调控血管活性物质的表达、防止心肌细胞肥大从而防治左室肥厚、保护肾脏功能等作用。临床实践依据及基础实验依据皆表明降压通脉方在对高血压靶器官的保护方面有很大的临床发展优势与潜力,但目前对于该方的药效作用物质基础与机制阐释尚不明确,需要进一步研究与挖掘。
网络药理学基于人体是一个由众多基因或其产物相互作用从而构成的一个复杂的生物网络,网络的失衡是疾病发生的本质[13]的理念,提出运用网络分析的方法分析药物、疾病及靶点间复杂的相互作用关系[14],可以更好的揭示非单一靶点的药物的作用特点[15]。网络药理学对疾病本质的认识与中医“以平为期”的治疗观点基本吻合。运用网络药理学的方法研究复方中药可以揭示药物与作用靶点间的相互关系,阐释中药活性成分间的协同作用机制,确证中药药效的微观生物学基础[16]。
基于以上分析,本研究基于网络药理学方法,对降压通脉方的预测靶点进行相关网络与生物功能分析,从而加深对该方的作用机制的认识与理解,为进一步的实验验证及临床拓展应用提供依据。
1 材料与方法
1.1 材料 中医药百科全书数据库ETCM(http://www.nrc.ac.cn:9090/ETCM/);BATMAN-TCM在线分析工具(http://bionet.ncpsb.org/BATMAN-TCM-tcm/);STRING在线数据库(https://string-db.org/);Cytoscape软件(版本3.7.2)及CytoNCA、cytohubba、MCODE、Enrichment Map及AutoAnnotate插件;David 6.8生物信息学分析数据库(https://david.ncifcrf.gov);R语言(版本3.6.1)。
1.2 降压通脉方活性成分的筛选及靶点的预测 降压通脉方由丹参、瓜蒌、红花、郁金、鸡血藤、薤白、香附、黄芩、菊花、草决明及珍珠母共11味药物组成。其中珍珠母运用BATMAN-TCM在线分析工具获取活性成分及靶点,其余10味药物运用ETCM数据库获取活性成分及预测靶标。
BATMAN-TCM[17](http://bionet.ncpsb.org/BATMAN-TCM-tcm/)是一个在线生物信息学分析工具,具有成分靶点预测、生物功能分析等功能。本研究以“珍珠母”为关键词检索BATMAN-TCM数据库,以得分≥20與P<0.05为临界值,分别进行化合物筛选及作用靶点预测,并删去未预测到靶点的化合物。
ETCM数据库[18]是由中国工程院院士黄璐琦研究员带领中国中医科学院中药研究所、中药资源中心、北京大学中药学院等单位共同打造的具有自主知识产权的在线开源中医药数据平台,采用Pipeline Pilot ADMET、QED、MedChem Studio(3.0版)等多种权威算法计算药物动力学参数、药物相似度及预测作用靶点,并给出Tanimoto评分>0.8分的高相关性作用靶点。本研究运用ETCM检索降压通脉方中除珍珠母外的10味药物的活性成分及预测靶标,并删去未预测到靶点的化合物。
整合以上检索结果,最终得到降压通脉方的活性成分及潜在靶点。
1.3 “药物-活性成分-靶点”相互作用网络的构建与分析 将“1.2”中筛选出的活性成分与潜在靶点数据导入Cytoscape 3.7.2软件构建“药物-活性成分-靶点”相互作用网络。利用Cytoscape 3.7.2软件的Network Analyzer工具及CytoNCA插件计算网络拓扑参数,得到上述网络关系中的自由度(Degree)、介度中心性、(Betweeness Centrality,BC)、接近中心性(Closeness Centrality,CC)、特征向量中心性(Eigenvector Centrality,EC)等指标。筛选标准:1)DC值大于所有节点DC值中位数2倍的重要节点;2)其他几个指标大于所有节点中位数的节点,即得到关键节点,构成Hub节点靶标基因集,简称Hub集。
1.4 降压通脉方蛋白相互作用网络构建与分析 String数据库[19](http://string-db.org/cgi/input.pl,Version:11.0)是目前公认的查询下载蛋白相互作用关系的在线数据库,其收集了大量被实验证实及预测得到的蛋白相互作用关系的数据,共涉及24.6百万个蛋白,>2 000百万个相互作用。采用STRING在线数据库查询下载蛋白相互作用网络(PPI)数据(得分>0.9),结果导入Cytoscape 3.7.2软件绘制相互作用网络。利用Cytoscape 3.7.2软件的cytohubba插件选用最大团中心性(Maximal Clique Centrality,MCC)[20]计算方法获得得分前100位的关键靶标,构成MCC关键节点靶标基因集,简称MCC集。
作为一个复杂的自适应系统,生物网络由一组相互作用的单元“模块”组成,这些单元被认为是最小的功能实体[21]。探索这“模块”以及它们如何协同工作来执行复杂的功能导致表型改变,可能是阐明这种多成分中药复方的药理机制的一个很有希望的机会[22]。MCODE是cytoscape中一个基于聚类算法的插件,它可以根据拓扑结构对给定的网络进行聚类,找到密切联系的功能“模块”。其优势在于具有一种定向模式,允许在不考虑网络其余部分的情况下对感兴趣的簇进行微调,并允许检查与蛋白质网络相关的簇互连性[23]。利用Cytoscape 3.7.2软件的MCODE插件进行关键模块的识别,参数设置采用默认值。
1.5 基因功能和信号通路分析 使用DAVID在线分析工具对Hub集、MCC集及模块进行GO及KEGG Pathways分析。通过统计学P值和FDR值来评估富集结果,其中GO分析分为生物过程(Biological Process,BP),分子功能(Molecular Function,MF)和细胞组成(Cellular Component,CC)3大模块进行基因功能注释。保存结果后,阈值设置为Pvalue<0.01,FDR<0.01进行显著性富集结果筛选。对模块GO分析结果运用Cytoscape 3.7.2软件的Enrichment Map插件[24]进行网络构建及分析,对其中代表主要生物学主题的类似通路的簇使用AutoAnnotate插件,一个自动运行clusterMaker2以生成群集并根据所选属性的单词频率自动生成集群标签的插件,自动集群及定义其生物学主题。
2 结果
2.1 降压通脉方活性成分的筛选及靶标预测 在ETCM数据库中,分别以“丹参”“瓜蒌、“红花”“郁金”“鸡血藤”“薤白”“香附”“黄芩”“菊花”及“决明子”为关键词进行检索,整合检索结果,并删去未得到高相关性的靶点靶点的化合物,最终结果显示(截止到2019年7月):共筛选出成分224种,其中丹参34个,红花20个,鸡血藤5个,决明子24个,菊花10个,黄芩51个,薤白21个,香附11个、郁金5个,瓜蒌43个,去掉其中重复的成分13种,共211种;共预测到靶点602个。
通过BATMAN-TCM工具检索到珍珠母成分共8种,为铝、锌、铜、锰、铁、硅、碳酸钙、锶,其中3个化合物锌、锰、锶未通过BATMAN-TCM在线工具预测到得分≥20的靶点,其余5个化合物共得到靶点32个,重复的靶点26个。
综上所述,降压通脉方全方共筛选出成分216种,预测到靶点608个。
2.2 “药物-活性成分-靶点”相互作用网络分析 通过Cytoscape 3.7.2软件绘制降压通脉方的“药物-活性成分-靶点”相互作用网络图。见图1。图中正多边形代表药物,菱形代表活性成分,圆形代表作用靶点,且具有相互作用关系的节点间用直线连接,节点颜色的深浅与度值相对应。此网络结构中包含了835个节点和11 411条边,835个节点中包括11味药物、216种活性成分及608个作用靶点。图中药物所包含的成分成方阵排列在对应药物下方,而两味或多味药物共有的8个成分单独成行排列,其中丹参和黄芩共有的成分为黄芩苷和豆甾醇,红花和黄芩共有的成分为棕榈酸,红花和瓜蒌共有的成分为月桂酸、亚麻酸及硬脂酸,丹参、红花及黄芩三味药共有的成分为β-谷甾醇。对该网络的拓扑学性质进行分析(见表1)及筛选,筛选策略为:Degree>18且EC>0.01且BC>21.61且CC>0.28,共发现关键节点230个(见图2)。其中药物5味,按度值降序依次为黄芩、瓜蒌、丹参、薤白及红花;成分112个(见表2,仅提供度值排名前20位的成分信息);靶标113个,构成Hub靶标集(简称Hub集)。
2.3 降压通脉方蛋白相互作用网络构建与分析 蛋白相互作用网络结果詳见图2,图中节点表示蛋白,边表示蛋白之间的关联,节点颜色的深浅与度值相对应。图中共涉及457个节点,1 900条边,其中有160个靶标包括1.3中筛选出的113个靶点中的25个(SLC22A1、SLC22A3、SLC22A8、CYP3A43、SLC22A7、SERPINA6、SLC10A6、CYP27A1、IGHG1、ATP1A1、SLCO2B1、SLCO4A1、SLCO1B3、SLCO1C1、GLTP、SLC22A6、SLC16A1、PMP2、SLCO2A1、SEC14L2、SLC22A11、NQO2、GPER、PIM1、UGT3A1)未在数据库中找到与其他蛋白相互作用评分>0.9的相互作用关系,这160个蛋白不在相互作用网络中体现,另MT-CO2、MT-ATP6、MT-CO1、SLC51B、NOS2、ENSG00000196689、GPER1、PTGDR2、SLC51A不在预测到的靶标蛋白序列中,通过STRING数据库补充。使用cytohubba插件中MCC方法计算后筛选出得分排在前100的靶点,构成MCC靶标集(简称MCC集),结果如图3所示。
利用MCODE进行模块识别,得到29个模块。其中最大的模块为模块6,包含38个靶标,147个相互作用关系,评分7.946分;评分最高的模块为模块1,包含15个靶标,105个相互作用关系,评分15分;评分最低的模块为模块29,包含4个靶标,4个相互作用关系,评分2.667分。详见表3(仅提供评分>5分的模块信息)。
29个模块共涉及靶点247个,其中48个靶点与Hub集重叠,99个靶点与MCC集重叠,3种方法共同筛选出的靶点有22个,具体包括:CYP3A4、CYP3A5、CYP3A7、GABRB3、CYP1A1、RXRA、CYP2E1、CYP1A2、CYP2B6、CYP19A1、GABRA2、GABRA1、GABRA4、GABRA3、CYP2C9、GABRA6、GABRA5、CYP2C19、SULT2B1、AKR1C3、AKR1C1、CYP2A6。
2.4 基因功能和信号通路分析 使用DAVID数据库对Hub集、MCC集及模块涉及的靶标基因进行GO及KEGG Pathways富集分析,阈值设置为Pvalue<0.01,FDR<0.01。
通过GO富集分析发现,Hub集显著富集在30个BP、23个MF和10个CC中,MCC集显著富集在28个BP、28个MF和20个CC中。见图4。图4中横坐标为富集的Enrichment值,纵坐标为-log10PValue,圆点表示Hub集富集到的GO基因注释信息,三角形表示MCC集富集到的基因注释信息,图形的颜色与大小由FDR值和相关联的基因数量决定。其中Hub集主要富集在类固醇代谢过程、氧化还原、阴离子运输、一元羧酸运输、药物代谢过程、有机阴离子转运、对有机物的反应、对营养水平的反应、离子输运、维生素D代谢过程等生物学过程,膜组分、不溶性组分、细胞碎片、微粒体、泡状部分、外模、氯通道络合物、内质网膜、内质网、核膜-内质网等细胞组分及脂质结合、血红素结合、以还原黄素为组分的氧化还原酶活性、四吡咯结合、类固醇结合、芳香化酶活性、配体依赖性核受体活性、类固醇激素受体、有机阴离子跨膜转运活性、电子载流子活性等分子功能上;MCC集主要富集在氧化还原、γ-氨基丁酸信号途径、类固醇代谢过程、甾体生物合成过程、翻译延伸、细胞激素代谢过程、G蛋白偶联受体蛋白信号途径、第二信使介导的信号转导、药物代谢过程、激素生物合成过程等生物学过程,突触后膜、微粒体、泡状部分、氯通道络合物、突触部分、大核糖体亚单位、突触、细胞器膜、内质网膜、核膜-内质网等细胞组分及GABA受体活性、GABA-A受体活性、血红素结合、四吡咯结合、电子载流子活性、以还原的黄色蛋白为组分的氧化还原酶活性、氧结合、铁离子结合、类固醇脱氢酶活性、细胞外配体门控离子通道活性等分子功能上。两者共同富集到的基因注释信息有26个,包块类固醇代谢过程、氧化还原、药物代谢过程、细胞激素代谢过程、激素代谢过程、激素水平的调节剂及γ-氨基丁酸信号途径7个生物学过程,膜组分、不溶性组分、细胞碎片、微粒体、泡状部分、氯通道络合物、内质网膜、内质网、核膜-内质网9个细胞组分及血红素结合、以还原的黄色蛋白为组分的氧化还原酶活性、四吡咯结合、芳香化酶活性、电子载流子活性、氧结合、阴离子跨膜转运蛋白活性、铁离子结合、GABA-A受体活性、GABA受体活性10个分子功能。
对MCODE中评分>5的9个模块分别进行GO富集分析,共得到101个显著富集的生物学过程、54个显著富集的细胞组分及81个显著富集的分子功能,共236个富集结果,其中有38个重复值。对这些结果运用Cytoscape 3.7.2软件的Enrichment Map插件[24]及AutoAnnotate插件进行集群分析,Enrichment Map插件参数设置Node Cutoff Q-value<0.01,Edge Cutoff(Similarity)>0.375。见图5。图5中包含198个节点,2 723条边,共得到11个功能集群,其中最大的是离子跨膜转运及信号转导功能相关集群,共包含80个节点、1 773条边,最小的是类固醇脱氢酶活性和营养水平刺激相关功能集群,各包含2个节点,1条边。
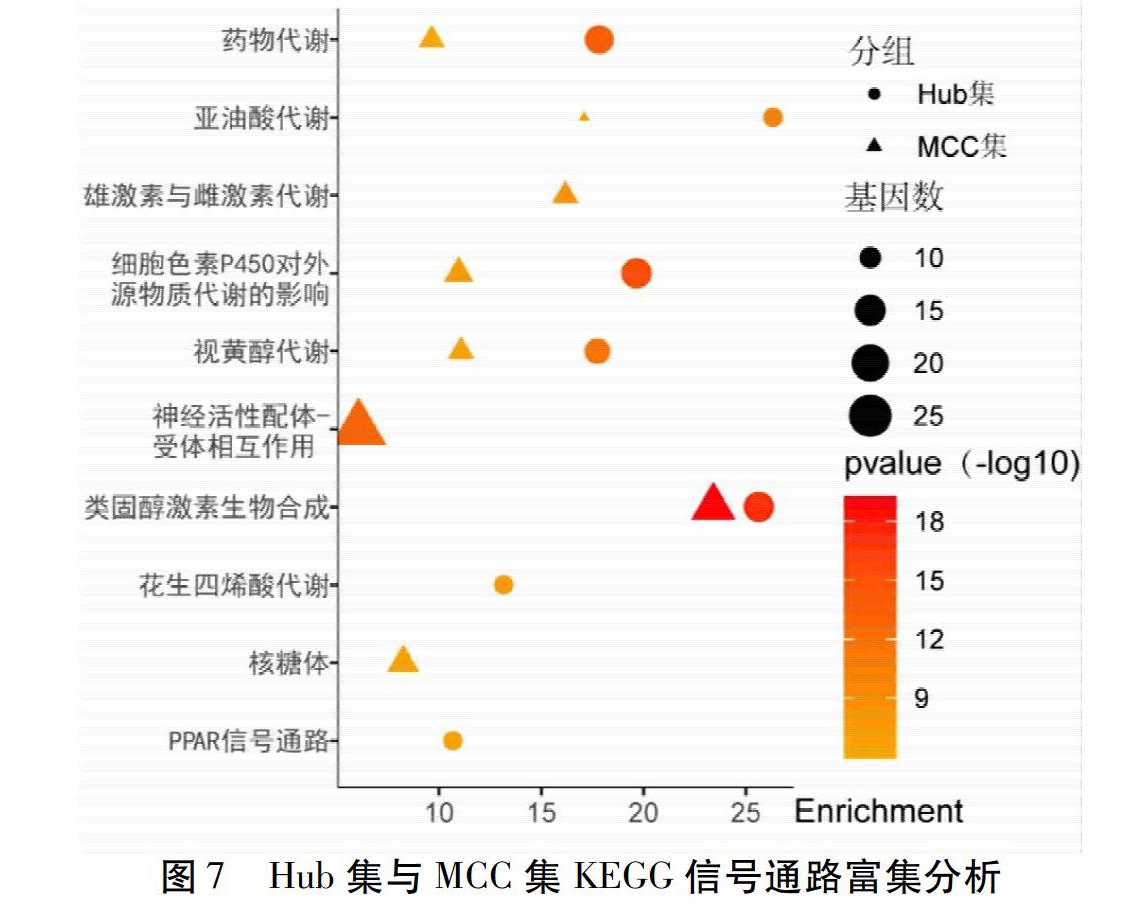
此外对Hub集和MCC集进行KEGG分析共得到10条信号通路。见图7。图7中圆点圆点表示Hub集富集到的KEGG信号通路,三角形表示MCC集富集到的基因注释信息,图形的颜色与大小由Pvalue和相关联的基因数量决定。对MCODE中评分>5的9个模块进行KEGG分析共得到17条信号通路,详见图6。
3 讨论
中医复方中各药物之间不是简单的叠加与堆砌,而是相须相使等通过配伍有机的结合成一个整体,其作用机制是单一成分或靶点难以解释的。网络药理学是一种用联系的理念看待生物体,结合生物信息学技术和网络分析、药理学等的方法来阐释靶标网络的协同效应和潜在机制的方法。因其与中医复方特点与理念高度契合,成为目前较为热门的中医复方研究方法。降压通脉方是名老中医效方,在对高血压靶器官的保护方面有良好的应用前景。进一步研究该方的药效作用物质基础与机制,有助于加深对该方的作用机制的认识与理解,为进一步的实验验证及临床拓展应用提供依据。
本研究通过网络药理学理念和技术,从降压通脉方全方11味药物中筛选出成分216个,预测到靶点608个。通过分析以这些成分和靶点构建的网络模型,共筛选出112个关键成分,这些成分中连接度前3名的化合物为β-谷甾醇(Β-Sitosterol)、泻根醇酸(Bryonolic Acid)及月桂酸(Lauric Acid)。其中β-谷甾醇是一种常见的植物甾醇,有广泛的药理作用,如抗氧化、抗高血脂、抗炎、免疫调节、抗肿瘤等,特别是防治心脑血管疾病、降低胆固醇等方面备受关注[25]。泻根醇酸是一种四环三萜类化合物,具有细胞毒性、抗炎、抗肿瘤、抗氧化等生物活性[26]。月桂酸是典型的中链脂肪酸,研究表明[27]可抑制高脂饲喂小鼠脂肪沉积,并在一定程度上缓解小鼠体内的炎性反应。网络中连接度排名前三的靶点为CYP3A4、ABCB1和ABCG2。CYP3A4中文名为细胞色素P450 3A4酶,是细胞色素P450氧化酶家族成员之一,是药物在体内代谢的重要影响因素之一。ABCB1中文名为P-糖蛋白、ABCG2中文名为乳腺癌耐药蛋白,两者均是膜转运体ABC超家族的成员,是具有ATP结合区域的苦熬膜蛋白,通过主动运输的方式转运多种物质包括药物,是药物耐受的重要相关蛋白[28]。
蛋白质相互作用网络结果显示降压通脉方的靶点间存在着复杂的相互作用关系,其中联系更为密切的构成模块单元发挥作用。经过计算,ANXA1、ADCY1、CHRM2、CNR1、CNR2、HTR1A、ADORA1、PTGER3、PTGDR2、GABBR1、GABBR2、GPER1、SUCNR1、P2RY12、OPRK1等可能是降压通脉方发挥作用的主要靶蛋白。此外Hub集是通过药物成分与靶点间的互作关系经计算得到的,一定意义上可以看做是复方药物直接影响的靶点,MCC集是通过靶点间的相互作用关系计算得到的,一定意义上代表复方药物对复杂的生物网络起调节作用的关键靶点。通过比较Hub集与MCC集,有22个靶标蛋白相重叠,说明一部分药物直接作用的蛋白在机体调控中起到重要作用,但更多的药物作用是通过靶点间的相互作用实现的。
通过富集分析可知,降压通脉方的靶基因主要富集在类固醇代谢过程、氧化还原、药物代谢过程、细胞激素代谢过程、激素代谢过程、激素水平的调节剂及γ-氨基丁酸信号途径等生物学过程,这些生物学过程与免疫、炎性反应、脂代谢等高度相关,可能是其作用广泛的物质基础。KEGG通路分析筛选出10条信号通路,主要包括机体内重要的小分子物资代谢通路:亚油酸、视黄醇、花生四烯酸及类固醇激素等,这些物质及代谢产物在机体内发挥重要作用,如在炎性反应、免疫反应、糖脂代谢等方面,而这些方面在心脑血管疾病、肾脏病等高血压易损伤的靶器官的疾病发生发展过程中均起到重要的作用,提示该方在上述疾病治疗中的潜力。此外还涉及到信号传导的相关通路2条,分别为神经活性配体-受体相互作用通路、PPAR信号通路。其中PPAR信号通路中PPARs是与维甲酸、类固醇和甲状腺激素相关的配体激活转录因子超家族核激素受体成员,主要调节脂肪代谢酶的转录,从而调节能量代谢、生长发育等生物功能,还参与细胞生长、分化与凋亡的调节。
通过模块分析可知,大多数模块的生物功能相对集中,仅涉及1~2个方面,复杂的中药复方通过作用靶标间的相互作用形成一个个功能模块,这些模块互相影响与调节,协调发挥作用。对其中得分较高的9个模块进行KEGG信号通路分析,结果发现其除与Hub集及MCC集共同涉及的神经活性配体-受体相互作用、核糖体、类固醇激素生物合成、雄激素与雌激素代谢、氧化磷酸化、PPAR信号通路等信号通路外,还涉及心肌收缩、帕金森病、阿尔茨海默病、肌痿侧索硬化症等疾病的信号通路,此外还涉及糖脂代谢的信号通路:脂肪细胞因子信号通路及胰岛素信号通路,与复制和转录相关的长期增益效应等及与能量代谢相关的柠檬酸循环。
综上所述,本研究以名中医效方降压通脉方为研究对象,通过网络药理学相关理念和方法分析降压通脉方作用机制。研究表明此方涉及多种信号通路和生物过程,體现了“多成分-多靶点-多途径”的协同作用特点,为进一步深入研究降压通脉方药理作用奠定基础。
参考文献
[1]焦东海.郭士魁老中医冠心病Ⅱ号方创立依据及经验方[J].中成药,1990,12(3):23-24.
[2]王亚红,肖文君,罗斯琼,等.降压通脉方对自发性高血压大鼠血压及左心室肥厚的影响[J].中华中医药杂志,2010,25(3):369-371.
[3]王琛.降压通脉方逆转高血压左室肥厚的信号转导通路机制初探[D].北京:北京中医药大学,2009.
[4]傅强.降压通脉方对左室肥厚高血压患者的临床及血管活性物质改变的观察[J].云南中医中药杂志,2004,25(4):15-17.
[5]王亚红.降压通脉方对自发性高血压大鼠血压及左心室肥厚的影响及对信号转导通路蛋白表达的影响[C].南昌:第十次中国中西医结合学会心血管病学术大会暨第五次江西省中西医结合学会心血管病学术大会论文汇编,2010.
[6]王亚红.降压通脉方对自发性高血压大鼠血压及左心室肥厚的影响[C].西宁:第四届全国血脂分析与临床学术研讨会暨第九届全国脂蛋白学术会议论文汇编,2008.
[7]王亚红.降压通脉方对自发性高血压大鼠血压及左心室肥厚的影响[C].上海:第一届全国中西医结合心血管病中青年医师论坛论文汇编,2008.
[8]曹征.降压通脉方对自发性高血压大鼠肾脏损害的实验及机理研究[D].北京:北京中医药大学,2007.
[9]罗斯琼,王亚红,王琛.降压通脉方对血管紧张素Ⅱ诱导心肌细胞肥大的影响[J].中国中医基础医学杂志,2009,15(8):584-586.
[10]王硕,王硕仁,赵悦茹,等.降压通脉方对高血压病左室肥厚患者血管活性物质的影响[J].中国中西医结合杂志,2002,22(4):274-276.
[11]秦建国.降压通脉方对高血压病肾损害大鼠尿微量白蛋白、NAG酶的调节作用[C].北京:中华中医药学会第二十一届全国中医肾病学术会议论文汇编,2008.
[12]罗斯琼.活血镇心法逆转高血压左室肥厚理论及作用机制初探[D].北京:北京中医药大学,2008.
[13]Tang F,Tang Q,Tian Y,et al.Network pharmacology-based prediction of the active ingredients and potential targets of Mahuang Fuzi Xixin decoction for application to allergic rhinitis[J].Journal of Ethnopharmacology,2015,176(Complete):402-412.
[14]Hopkins A L.Network pharmacology[J].Nature Biotechnology,2007,25(10):1110-1111.
[15]Hong H,Feuers R,Tang K,et al.Drug Repositioning Through Network Pharmacology[J].Current Topics in Medicinal Chemistry,2016,16(30):3646-3656.
[16]解静,高杉,李琳,等.网络药理学在中药领域中的研究进展与应用策略[J].中草药,2019,50(10):2257-2265.
[17]Zhongyang L,Feifei G,Yong W,et al.BATMAN-TCM:a Bioinformatics Analysis Tool for Molecular mechANism of Traditional Chinese Medicine[J].Scientific Reports,2016,16:6.
[18]Hai-Yu X,Yan-Qiong Z,Zhen-Ming L,et al.ETCM:an encyclopaedia of traditional Chinese medicine[J].Nucleic Acids Research,2019,47(D1):976-982.
[19]von MC.,Jensen LJ,Snel B,et al.STRING:known and predicted protein-protein associations,integrated and transferred across organisms[J].Nucleic Acids Research,2005,33(Database issue):D433-D437.
[20]Meghanathan N.Maximal Clique Size Versus Centrality[C].Springer India:A Correlation Analysis for Complex Real-World Network Graphs,New Delhi,2016.
[21]Sales-Pardo,Marta.The importance of being modular[J].Science,2017,357(6347):128-129.
[22]Wang P,Dai L,Zhou W,et al.Intermodule Coupling Analysis of Huang-Lian-Jie-Du Decoction on Stroke[J].Frontiers in Pharmacology,2019,10:1288.
[23]Bader G D,Hogue C W.An automated method for finding molecular complexes in large protein interaction networks[J].BMC Bioinformatics,2003,4(1):2.
[24]Daniele M,Ruth I,Oliver S,et al.Enrichment Map:A Network-Based Method for Gene-Set Enrichment Visualization and Interpretation[J].Plos One,2010,5(11):e13984.
[25]刘威良,姬昱,黄艾祥.β-谷甾醇的研究及开发进展[J].农产品加工,2019,18(1):83-85.
[26]张胜男,李煌,张玉琴,等.泻根醇酸对NMDA诱导的PC12细胞损伤的保护作用研究[J].天然产物研究与开发,2015,139(1):139-142,162.
[27]丁同庆,张枫琳,黄晓刚,等.月桂酸对高脂日粮饲喂小鼠脂肪沉积及炎症反应的影响[J].养殖与饲料,2019,18(11):28-31.
[28]李君蕊,付盈菊,赵宝春,等.ABCB1和ABCG2基因多态性与急性药物中毒性脑病的相关研究[J].中國急救医学,2014,34(3):233-237.
(2020-01-08收稿 责任编辑:苍宁)
