聚乳酸/聚酯酰胺复合纳米纤维膜制备及性能
2020-06-22崔心想王艳贺韩立彬孙奇浩宋晓峰
崔心想,王艳贺,韩立彬,孙奇浩,宋晓峰
(长春工业大学化学工程学院,吉林长春 130012)
随着石油资源的日益消耗和低碳环保意识的增强,生物资源的利用和开发引起了人们的广泛关注。饱和族脂肪族聚酯[1],尤其聚乳酸(PLA)是研究最成熟的生物可降解聚合物,主要应用于生物医学和生态环境等相关领域,但是由于其较低的热学和力学性能而限制了应用范围。
近年来,研究寿命可控、生物相容、生物可降解、物理力学性能优良的聚合物去服务于医疗和环保等领域成为当前科学界的目标之一。脂肪族聚酰胺具有良好的热学和力学性能,由于其在体内降解非常缓慢,通常被认为是不可生物降解的材料。聚酯酰胺(PEA)是指分子主链上含有酯键和酰胺键的聚合物。PEA 可以将聚酯的生物相容性和生物可降解性与聚酰胺的热学和力学性能相结合[2],并可通过调节酯键/酰胺键比达到灵活调节材料性能的目的。同时,含有α–氨基酸的PEA 提高了聚合物材料的生物学性能[3],如有助于更好的细胞聚合物相互作用,允许引入悬垂的活性基团,并改善聚合物材料的热塑性、降解性,容易被生物有机体新陈代谢或者排出体外。生物可降解PEA 已广泛应用于生物医学领域[4]。因此,将PEA 与PLA 复合成静电纺丝纳米纤维膜应用于生物组织工程支架及药物释放领域成为一个热门的研究方向。
目前,PEA 合成途径主要有:合成含有肽链结构的共聚物扩链连接[5],合成氨基酸二异氰酸酯再共聚,由氨基酸–N–羧基内酸酐(氨基酸NCAs)开环聚合[6],吗啉二酮开环聚合[7],内酰胺和内酯的开环共聚[8],二醇、二酸、二胺和氨基酸熔融反应[9],二酰胺二醇和二氯化物或二酯缩聚反应,二酯二胺盐与活化的二羧酸[10]或酰氯的反应[11]。陶友华[12]提出了利用氨基酸的成环形成内酰胺单体,再通过内酰胺开环聚合制备聚氨基酸的新方法。A. C. Fonseca 等[13]通过基于α–氨基酸的二胺和基于L–乳酸的二酰氯之间的界面聚合来制备PEA。Feng Y 等[14]介绍了吗啉 –2,5– 二酮衍生物的合成及其开环聚合PEA 的方法。
已经有很多文献探讨了PEA 的合成及其在生物医学领域的应用,但是通过乙二醇溶液聚合制备双端羟基大分子单体,经六亚甲基二异氰酸酯将合成端羟基的大分子单体扩链连接起来比较少见。因此,笔者使用六亚甲基二异氰酸酯将合成的端羟基大分子单体扩链连接合成新型PEA——主链中含有L–丙氨酸、乙二醇、己内酯的生物可降解聚合物[P–(CL–EA–CL)],并将其与 PLA 共混电纺成膜,以期获得生物相容性、降解性及力学和热性能良好的复合纳米纤维膜。
1 实验部分
1.1 原材料
左旋PLA (PLLA):数均分子量70 000,海正生物材料有限公司;
乙二醇、ε– 己内酯 (CL)、甲苯:分析纯,Aladdin 公司,使用前用CaH2进行减压蒸馏除水;
辛酸亚锡[Sn(Oct)2]:Aladdin 公司,使用脱水甲苯制备0.1 g/mL 的溶液;
L– 丙氨酸、对甲苯磺酸一水合物(TsOH·H2O)、六亚甲基二异氰酸酯(HDI)、二月桂酸二丁锡(DBTL):麦克林生化科技有限公司。
1.2 仪器及设备
傅里叶变换红外光谱(FTIR)仪:Nicolet iS10型,美国赛莫飞世尔有限公司;
核磁共振波谱仪 (NMR):Avance Ⅲ型,400 MHz,瑞士 Bruker 公司;
差示扫描量热(DSC)仪:DSC Q20 型,美国TA公司;
扫描电子显微镜 (SEM):JSM–7610F 型,日本Jeol 公司;
接触角测试仪:HARKE–SPCA 型,北京哈科试验仪器厂;
万能拉力机:YHS–229WG 型,上海益环仪器科技有限公司。
1.3 P–(CL–EA–CL)合成
第一步,首先合成L–丙氨酸–乙二醇–L–丙氨酸(EA)单体。在250 mL 三口烧瓶中依次加入搅 拌 子、0.01 mol 乙 二 醇 (0.556 4 mL)、0.02 mol L– 丙 氨 酸 (1.781 8 g)、0.02 mol TsOH ·H2O(3.804 2 g)、100 mL 甲苯。将三口烧瓶放入油浴锅中在130℃下回流反应24 h,对反应后的产物进行石油醚–异丙醇–石油醚沉淀纯化处理,放入冰箱中冷却过夜;最后将沉淀产物抽滤出来,在真空烘箱中干燥12 h 后得到EA 产物。
第二步,合成 CL–EA–CL 大分子单体。控制EA 与CL 的物质的量比即接枝比为1 ∶4,在100 mL 的磨口反应管中依次加入搅拌子、0.001 8 mol 的EA(1 g),然后将磨口反应管密封,反复抽真空充氮气三次,将瓶中充满氮气。使用注射器依次向磨口反应管中加入0.007 2 mol 己内酯 (0.813 5 ml)、1 mL Sn(Oct)2和 20 mL 纯 化 好的甲苯。将加好药品的磨口反应管放入油浴锅中,在120℃下反应24 h。对反应后的产物进行石油醚沉淀处理,放入冰箱中冷却过夜;最后将沉淀产物抽滤出来,在真空烘箱中干燥12 h 后得到CL–EA–CL–1 产物。控制 EA 与 CL 的物质的量比为1 ∶ 6 和 1 ∶ 8,使用同样方法合成 CL–EA–CL–2,CL–EA–CL–3 产物。
第三步,扩链 CL–EA–CL 单体合成 PEA。在100 mL 的磨口反应管中依次加入搅拌子、1 g的 CL–EA–CL–1,然后将磨口反应管密封,反复抽真空充氮气三次,将瓶中充满氮气。使用注射器依次向磨口反应管中加入0.159 4 mL HDI,0.299 2 mL DBTL 和20 mL 纯化好的甲苯[15]。将加好药品的磨口反应管放入油浴锅中,在95℃下反应12 h。对反应后的产物进行石油醚沉淀处理,放入冰箱中冷却过夜;最后将沉淀产物抽滤出来,在真空烘箱中干燥 12 h 后得到 P–(CL–EA–CL)–1 产物。使用同样方法合成 P–(CL–EA–CL)–2,P–(CL–EA–CL)–3 产物。
EA,CL–EA–CL,P–(CL–EA–CL)的合成路线见图1。

图 1 EA,CL–EA–CL,P–(CL–EA–CL)的合成路线
1.4 复合纳米纤维膜制备
将计算量的 P–(CL–EA–CL)–1,PLLA 分别加入0.5 mL 二甲基甲酰胺和5 mL 氯仿中配制成质量分数为8%的聚合物溶液,P–(CL–EA–CL)添加比例分别是10%,30%和50%。再向溶液中放入1%的苄基三乙基氯化铵来提高溶液的导电性。溶液搅拌过夜后静置1~2 h,直至没有气泡后将混合溶液倒入5 mL 的注射器中进行静电纺丝。纺丝条件:0.9 mm 的平头不锈钢针头,针头和铝箔接收板之间的距离为15 cm,电压为7 kV,注射速度为6 µL/min。将静电纺丝纤维膜放入真空干燥箱中30℃真空干燥24 h。采用同样方法制备出P–(CL–EA–CL)–2,P–(CL–EA–CL)–3 添加比例是 30% 的静电纺丝纤维膜。不同纤维膜基体配方见表1。

表1 不同纤维膜基体配方 %
1.5 性能测试与表征
FTIR 分析:将样品与溴化钾粉末混合研磨压片,在常温下选用的波长范围为400~4 000 cm–1。
核磁共振质谱(1H NMR)分析:室温下以氘代二甲基亚砜(d-DMSO)为溶剂,使用四甲基硅烷作为内标对比物。
DSC 分析:在 N2保护下以 10℃/min 的升降温速率在0~200℃的范围内加热和冷却,样品质量5~10 mg。
微观形貌分析:采用SEM 放大5 000 倍来观察,在测试之前进行表面喷金处理。
接触角测试:将纤维膜裁成矩形,通过接触角测试仪测定接触角,每个样品测试5 个平行样。
拉伸性能测试:在室温下使用万能拉力机对共混纤维膜进行拉力测试。样品裁剪成10 mm×40 mm 的长方形样条,拉伸速率5 mm/min,每个样品膜测试5 次取平均值。
降解测试:将共混纤维膜放入带盖的50 mL 0.1 M 的NaOH 玻璃瓶内,密封后固定在恒温振荡箱内振荡降解,控制降解温度为37℃,选择不同的时间间隔取出样品,使用蒸馏水彻底清洗后干燥称重。每个共混膜同时设计5 组平行测试。同时采用0.1 M 的H2SO4溶液代替NaOH 溶液同步进行降解测试,纯的PLLA 纤维膜被用来作为对比样。
细胞毒性测试:将纤维膜切好放入24 孔的培养皿中进行培养,取细胞培养液放在一个单独的培养皿中进行阴性对照。通过MTT 法检测小鼠体内细胞在纤维膜中的活力,并使用酶联免疫检测仪进行测量。增殖存活率(RGR)=检测的吸光度/对照样吸光度 ×100%。根据 ISO 10993–5,当RGR>75%时就证明材料不存在潜在的细胞毒性。
2 结果和讨论
2.1 结构分析
EA,CL–EA–CL 和 P–(CL–EA–CL) 的 红 外谱图见图2。EA 中酯基—C=O 的吸收峰出现在1 750 cm–1处。CL–EA–CL 中 —C=O 的 吸 收 峰向左偏移出现在 1 724 cm–1处,1 190 cm–1处是酯基上 C—O 的伸缩振动峰,2 945 cm–1处是亚甲基上C—H 的伸缩振动峰,同时出现了己内酯链末端羟基的特征峰3 437 cm–1和五个相连的亚甲基C—H 的伸缩振动峰732 cm–1,说明接枝产物成功合成。P–(CL–EA–CL)中—C=O 的吸收峰出现在1 725 cm–1处,1 623 cm–1处为新出现的酯酰胺特征峰。红外谱图证明了由HDI 扩链的P–(CL–EA–CL)产物成功合成。

图 2 EA,CL–EA–CL,P–(CL–EA–CL)的红外谱图
EA,CL–EA–CL 和 P–(CL–EA–CL)的核磁共振谱图见图3。由图3a 可看出,EA 的谱图中,在δ=2.29(a)处出现的是与苯环相连的甲基峰(C—CH3),在δ=7.13(b)和δ=7.48(c)处是苯环上的亚甲基峰(—CH2)。在δ=1.39(d)处出现了丙氨酸侧链上的甲基峰,δ=4.12(e)处为丙氨酸上与酯基和氨基相连的次甲基峰(—CH),δ=4.39(f)处为乙二醇上的亚甲基峰。由图 3b 可以看出,CL–EA–CL 的谱图中,在PCL 链中的亚甲基峰出现在δ=1.29(h)和δ=1.53(i)处,在δ=2.19(j)和δ=3.98(g)处出现的分别是与酰胺键和酯键相连的亚甲基峰。由图3c 可看出,P–(CL–EA–CL)的谱图中,在 HDI 链中的亚甲基峰出现在δ=1.27(m)和δ=1.50(l)处,在δ=2.78(k)处出现的是与酯酰胺键相连的亚甲基峰。核磁谱图清晰说明了EA 的合成及其成功接枝上CL,并在HDI 的作用下进行均聚扩链成 P–(CL–EA–CL)。
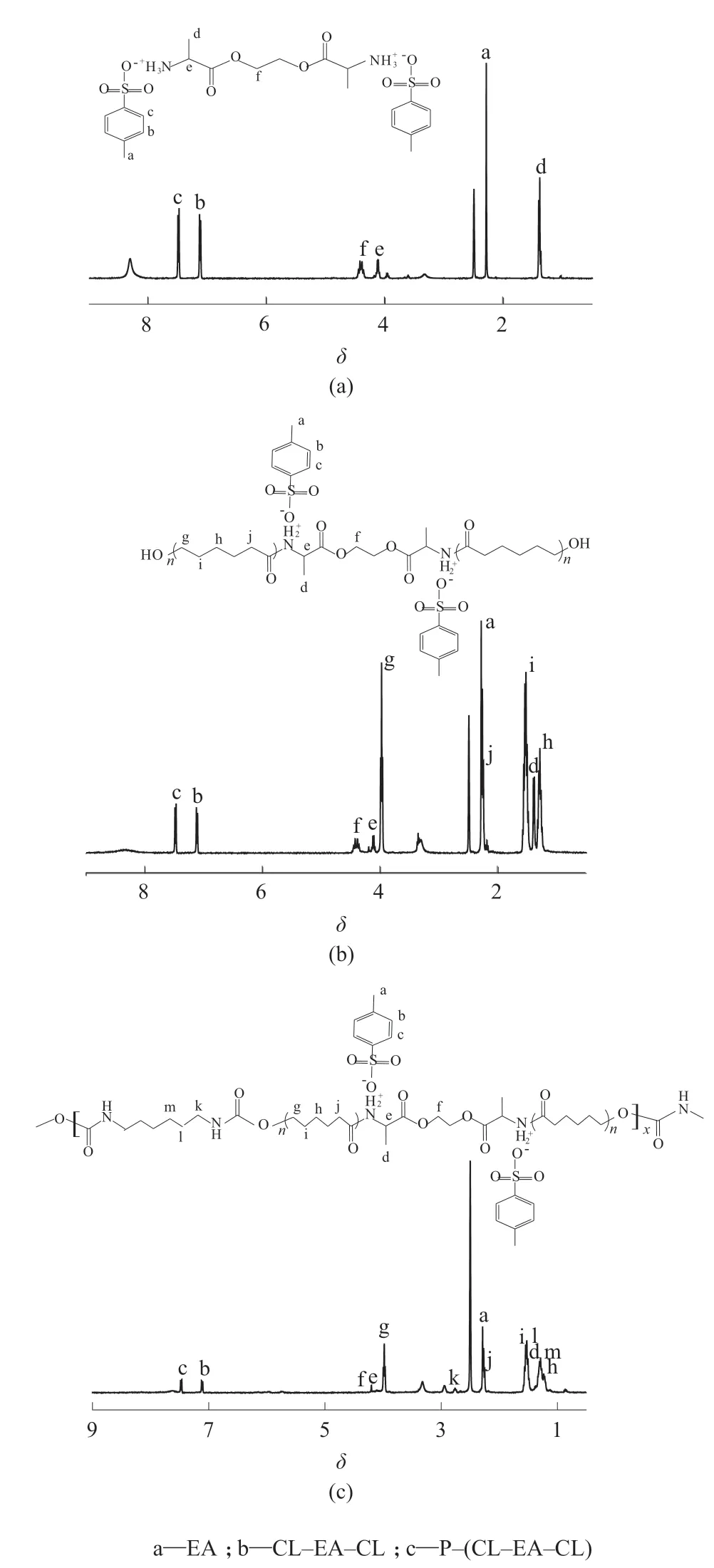
图 3 EA,CL–EA–CL,P–(CL–EA–CL)的核磁谱图
2.2 热力学行为
图4 给出了纯PLLA 纤维膜与不同共混比PLLA/P–(CL–EA–CL)–1 纤维膜 的 第二 次 升温DSC 曲线,相应热力学数据见表2,其中Tg为玻璃化转变温度;Tc为结晶温度;Tm为熔融温度;ΔHm为熔融晗;Xc为结晶度。

图4 纯PLLA 纤维膜与不同共混比纤维膜第二次升温DSC 曲线

表2 纯PLLA 纤维膜与不同共混比纤维膜的DSC 数据
从表2 可以看出,与纯PLLA 纤维膜相比较,PLLA/P–(CL–EA–CL)–1 共混纤维膜的Tg有所降低,表明其与PLLA 间有良好的界面粘结和相容性。
当 P–(CL–EA–CL)–1 比例从 0% 增加到 50%,共混膜中PLLA 的Tc从102.94℃降到95.31℃。这是由于 P–(CL–EA–CL)–1 的加入使得共混纤维膜中PLLA 由单一向的均质成核变成两相之间的异质成核,使得Xc有所降低和共混膜中的聚集态发生变化所致。
与纯 PLLA 纤维膜相比较,PLLA/P–(CL–EA–CL)–1 共混纤维膜的Xc有所降低。随着 P–(CL–EA–CL)–1 比例增加,Xc先增加后减小。当少量的P–(CL–EA–CL)–1 混 入 时,PLLA 的Xc从 28% 下降到19%,这说明PLLA 的结晶过程受到P–(CL–EA–CL)–1 的影响。少量的 P–(CL–EA–CL)–1 阻止了PLLA 分子链的整齐折叠和重新排列,从而导致了 PLLA 的Xc降低。适量 P–(CL–EA–CL)–1 的加入使得其与PLLA 相互渗透与缠结形成界面,促进PLLA 的异质结晶从而使得共混膜的结晶度略有提高。随着 P–(CL–EA–CL)–1 比例的增加,其在共混膜中趋向于相互聚集形成类晶体限制PLLA 分子量的运动排列,导致共混膜结晶度降低。
图5 给出了纯PLLA 与不同接枝比PLLA/P–(CL–EA–CL)纤维膜的第二次升温 DSC 曲线,相应热力学数据见表3。

图5 纯PLLA 纤维膜与不同接枝比共混膜第二次升温DSC 曲线

表3 纯PLLA 纤维膜与不同接枝比纤维膜的DSC 数据
从表3 可以看出,与纯PLLA 纤维膜相比较,PLLA/P–(CL–EA–CL)共混纤维膜的Tg有所降低,表明其与PLLA 之间有良好的界面粘结和相容性。当接枝比例为 1 ∶ 6 时,PLLA/P–(CL–EA–CL)–2共混纤维膜的Tg最小,表明 P–(CL–EA–CL)–2 与PLLA 的相容性最好。这是由于适量的CL 链段使得其与PLLA 分子链段之间更好的界面接触和粘结。
与纯PLLA 纤维膜相比较,共混纤维膜的Tc有所降低。当接枝比例从1 ∶4 增加到1 ∶8 时,共混膜中PLLA 的Tc先增加后减小。这是由于P–(CL–EA–CL)中 CL 链段的长短影响其与 PLLA的界面接触和粘结。较短或较长的CL 链段会造成P–(CL–EA–CL)与 PLLA 接触粘结较差和更容易自聚使得聚集态发生变化所致。
与纯PLLA 纤维膜相比较,共混纤维膜的Xc有所降低,PLLA 的Xc从28%下降到20%,这说明PLLA 的结晶过程受到 P–(CL–EA–CL)的影响。这是由于P–(CL–EA–CL)的加入使得其在共混膜中趋向于相互聚集形成类晶体限制PLLA 分子链的整齐折叠和重新排列,导致共混膜结晶度降低。共混膜Xc保持一致且不会随着接枝比例的增加而改变,这说明P–(CL–EA–CL)中CL 链段的长短对共混膜的Xc影响较小。
2.3 纤维膜形态
图6 是纯PLLA 及其共混纤维的SEM 照片。由图6 可看出,纯PLLA 纤维膜表面光滑,粗细比较均匀。共混纤维的直径略小于纯PLLA 纤维的直径,且表面粗糙程度和粗细不均。随着P–(CL–EA–CL)–1 的含量增加,共混纤维的表面较为粗糙且粗细分布不均。对比图6d~图6f 可以发现,P–(CL–EA–CL)添加量为30%时,不同接枝比条件下共混纤维的表面均较为光滑且粗细比较均匀,整体形态较好,适合应用到生物医学领域尤其是组织工程上。

图6 纯PLLA 及共混纤维膜的SEM 照片
2.4 浸润性
图7 示出不同共混比时纤维膜的水接触角。从图7 可以看出,纯PLLA 纤维膜的水接触角最大,为 138.2°,且随着 P–(CL–EA–CL)–1 含量的增加,共混纤维膜的接触角有较大幅度的减小。P–(CL–EA–CL)–1 少量添加时共混纤维膜的亲水性有略微改善。当 P–(CL–EA–CL)–1 的添加量达到 30%,50%时,共混纤维膜的亲水性得到大幅度的提高,且不会随着添加量的增加而改变。这是因为P–(CL–EA–CL)–1 具有亲水性,同时主链中的酰胺键也会为共混纤维膜内部带来氢键的作用[16]。

图7 不同共混比时纤维膜的水接触角
图8 是不同接枝比时纤维膜的水接触角。从图8 可以看出,PLLA/P–(CL–EA–CL)–1 共混纤维膜的接触角最小,为76.3°,随着接枝CL 比例的增加,共混纤维膜的水接触角略有增加,亲水性小幅降低,这是由于CL 链为疏水性造成的。
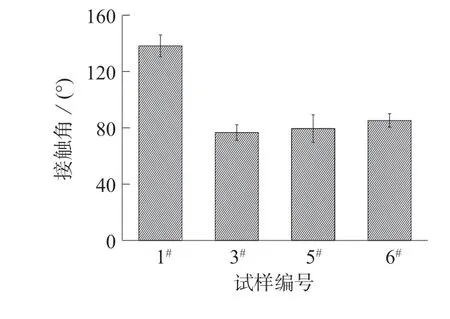
图8 不同接枝比时纤维膜的水接触角
2.5 力学性能
图9 示出不同共混比时纤维膜的拉伸性能。由图 9 可看出,随着 P–(CL–EA–CL)–1 含量的增加,拉伸强度先逐渐增大随后急剧减小,当含量为30%时达到最大值11.43 MPa,含量为50%时迅速降低到 6.27 MPa。断裂伸长率随着 P–(CL–EA–CL)–1含量的增加先基本不变而后急剧降低。
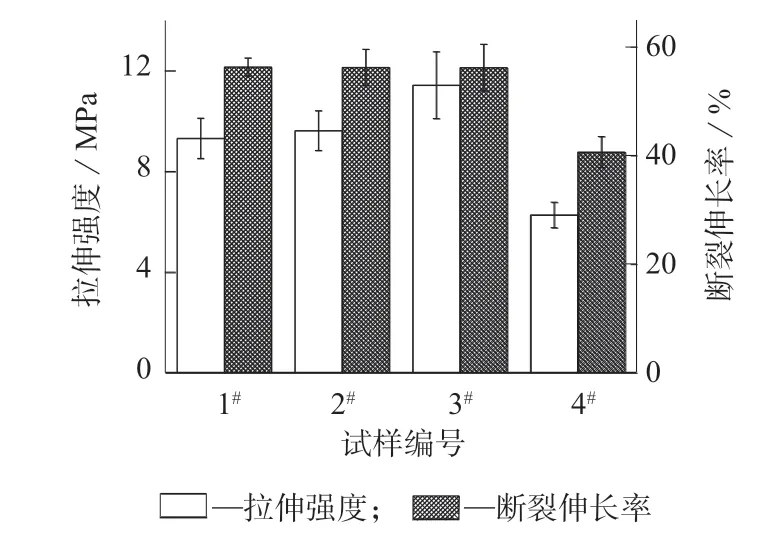
图9 不同共混比时纤维膜的拉伸性能
当 P–(CL–EA–CL)–1 含量较低时,其可在共混体系中良好地分散,可与纯PLLA 形成良好的界面粘附并为共混纤维膜带来拉伸强度的提升。当含量进一步增加时,P–(CL–EA–CL)–1 分散相会发生聚集而产生大的颗粒,导致其与PLLA 基质发生相分离,使得拉伸强度和断裂伸长率急剧下降。
图10 示出不同接枝比时纤维膜的拉伸性能。由图10 可看出,随着接枝比的增加,拉伸强度先增大后减小,当接枝比为1 ∶4 时达到最大值11.43 MPa,接枝比为1 ∶6 时迅速降低到6.9 MPa。

图10 不同接枝比时纤维膜的拉伸性能
断裂伸长率随着接枝比的增加略有提高。当接枝比例增加时,P–(CL–EA–CL)中 CL 链段更长且更具柔韧性,使得共混纤维膜受到外力时更好地转移到 P–(CL–EA–CL)上,断裂伸长率略有改善。与此同时,较长的CL 链段也使得共混纤维膜的拉伸强度有所降低。从图中可以看出,当接枝比为1 ∶4 时,共混纤维膜的拉伸强度和断裂伸长率较高。
图 11 给 出 了纯 PLLA 和 PLLA/P–(CL–EA–CL)–1(3#)共混膜的应力–应变曲线。从图11 可以看出,与纯PLLA 纤维膜相比,PLLA/P–(CL–EA–CL)–1 纤维具有更好的拉伸强度(从9.32 MPa提高到11.43 MPa),且在拉伸过程中形成均匀的细颈和稳定的应变硬化呈现出韧性断裂。这表明P–(CL–EA–CL)–1 对共混膜具有良好的增强效果。从组织工程的角度来看,这种材料可以有效改善共混纤维膜的力学性能,符合组织工程支架的应用条件。

图11 纯PLLA 和共混膜的应力–应变曲线
2.6 降解行为
图 12 给 出 了 纯 PLLA 膜 与 PLLA/P–(CL–EA–CL)–1(3#)共混膜在 H2SO4溶液和 NaOH 溶液中的失重曲线。纯 PLLA 膜与PLLA/P–(CL–EA–CL)–1 共混膜在不同酸碱度溶液中的降解速率和降解率是不同的,但失重曲线的趋势和失重率大致相同[17]。共混膜在H2SO4溶液(图12a)中降解7 d后,最终的失重率均在10%左右。这表明酸性环境对其降解的影响很小。然而共混膜在NaOH 溶液中降解速率较快,在72 h 和84 h 左右均表现出完全降解。这表明共混膜对碱性环境较敏感,并不是耐碱性的材料。P–(CL–EA–CL)–1 的加入略微延缓PLLA 在碱性中的降解速率。这是因为所合成的P–(CL–EA–CL)–1 具有亲水性和高化学稳定性。因此共混膜在生物组织工程领域更具使用价值。

图12 纯PLLA 膜与共混膜的失重曲线
2.7 细胞毒性
表 4 列出了纯 PLLA 纤维膜和 PLLA/P–(CL–EA–CL)–1(3#)共混纤维膜的RGR结果。纯 PLLA纤 维 膜 的RGR是 93.46%,PLLA/P–(CL–EA–CL)–1 共混纤维膜的RGR是 85.73%,两种纤维膜都表现为一级毒性。证明了 P–(CL–EA–CL)–1 聚酯酰胺具有良好的生物相容性,可以作为生物医药组织工程的候选者。

表4 纤维膜的细胞毒性
3 结论
通过溶液聚合方法首先合成EA 单体,然后通过EA 单体两端的氨基引发CL 开环聚合成CL–EA–CL 大分子单体,最后在 HDI 为扩链剂、DBTL 为催化剂下扩链成 P–(CL–EA–CL)产物。通过红外、核磁表征证明了产物的成功合成。将P–(CL–EA–CL)与 PLLA 共混电纺成膜,并对纤维膜进行DSC、接触角、SEM、力学性能、降解行为及细胞毒性表征。通过比较发现相比纯PLLA 纤维膜,P–(CL–EA–CL)–1 含量为 30% 的共混纤维膜的表面较为光滑且粗细比较均匀,整体形态较好、亲水性得到大幅度提高(76.3°)、力学性能有所增强,拉伸强度为11.43 MPa 且细胞毒性表现为一级毒性,具有良好的生物相容性。从组织工程的角度来看,这种材料具有较好的纤维形貌、力学性能、亲水性和生物相容性,符合组织工程支架的应用条件。
