气相色谱法测定初元复合肽营养饮品中氯丙醇含量
2020-06-22刘贤良李文贵沈震亚李岚
刘贤良 李文贵 沈震亚 李岚
(江中药业股份有限公司 江西南昌330096)
氯丙醇是氯原子取代丙三醇结构上羟基而成的一类化合物,主要有3-氯-1,2-丙二醇(3-MCPD)、2-氯-1,3-丙二醇(2-MCPD)、1,3-二氯-2-丙醇(1,3-DCP)及2,3-二氯-1-丙醇(2,3-DCP)。研究表明,氯丙醇存在肾脏毒性、生殖毒性、神经毒性、免疫毒性、致突变性等,且国际癌症研究机构认为它是一种非遗传性的可能致癌物[1]。自世界卫生组织(WHO)1993 年警告氯丙醇类物质存在严重毒性以来,食品中的氯丙醇污染正式成为国际食品安全问题之一,其相关食品安全研究随即也成为科研工作者广泛关注的热点。食品中的氯丙醇污染产生于酸水解植物蛋白(HVP)的生产过程,为提高氨基酸得率而加入过量的盐酸会使原料中存在的三酰甘油酯水解成丙三醇,并进一步反应生成氯丙醇。我国保健食品和婴幼儿食品不少采用酸水解蛋白为原料,随着酸水解植物蛋白常作为风味增强剂加入,其所含的3-MCPD 也相应增加,因此需按照GBT 5009.191-2006 测定方法(气相质谱法)对其进行氯丙醇监控,对于不具备质谱仪的企业来说,也可直接采用气相色谱法进行测定。因此本文采用较为广泛使用的气相法测定初元复合肽营养饮品中氯丙醇的含量,以提高对氯丙醇的质量监控能力。现报道如下:
1 仪器与试药
1.1 仪器 Agilent 7890 气相色谱仪;N-1100 旋转蒸发仪;H1650 离心机;SK-1 混匀器;梅特勒AG 245 电子天平。
1.2 试药 1,3-二氯-2-丙醇对照品(批号:C12481600,纯度97.5%),2,3-二氯-1-丙醇对照品(批号:CA12482000,纯度98.0%),均购于美国Dr-Ehrentorfer 公司;正己烷为重蒸试剂;乙酸乙酯、乙醚、无水硫酸钠(105 ℃烘烤3 h 后使用)、氯化钠、七氟丁酰基咪唑(西格玛公司),均为分析纯;水为超纯水。初元复合肽营养饮品为江中药业有限公司产品(批号:20170104、20180206、20180307、20180404、20180502、20180604)。
2 测定方法[2]
2.1 标准溶液的配制
2.1.1 标准品单标储备液的配制 取1,3-DCP 和2,3-DCP 对照品各约25 mg 分别置于25 ml 容量瓶中,精密称定,加入乙酸乙酯溶解并稀释至刻度,得到浓度均为1 000 μg/ml 的2 种氯丙醇单标储备液,并于4 ℃的冰箱中储存备用。
2.1.2 标准品工作溶液的配制 分别精密吸取以上2 种氯丙醇单标储备液各1 ml,置2 支10 ml 容量瓶中,加乙酸乙酯稀释至刻度,分别得到浓度均为100 μg/ml 的2 种氯丙醇单标工作溶液。从2 种氯丙醇的单标工作溶液精密吸取适量,置于同一25 ml容量瓶,加正己烷稀释至刻度,得到混合标准品工作溶液,2 种氯丙醇的浓度均为2 μg/ml。
2.2 样品溶液的制备 精密吸取样品溶液10 ml,加饱和氯化钠溶液(5 mol/L)6 g,超声处理15 min,用乙醚萃取3 次,每次15 ml,合并乙醚液并加无水硫酸钠3 g,静置10 min 后过滤,滤液于35 ℃下旋蒸至约2 ml,定量转移至5 ml 具塞试管中,加乙醚稀释至4 ml,加少量无水硫酸钠,振摇,静置15 min以上。
2.3 衍生化 精密量取试样溶液1 ml 于5 ml 具塞试管中,并于室温挥至近干,立即加入正己烷1 ml,用气密针加入七氟丁酰基咪唑50 μl,立即塞紧,涡旋混合后,70 ℃保温20 min。放冷至室温,加饱和氯化钠溶液3 ml,涡旋混合30 s,使两相分离,取有机相加无水硫酸钠约0.3 g 脱水,取溶液进行GC 测定。
2.4 空白试样制备 称取饱和氯化钠溶液6 g,同2.2 法净化、2.3 法衍生。
2.5 色谱条件[3~5]气相色谱仪(电子捕获检测器ECD)色谱条件:色谱柱,HP-5 二苯基甲基硅氧烷(25 m×0.32 mm×0.52 μm);载气为氮气(纯度>99.99%);流量为2.8 ml/min;色谱柱程序升温,50~100 ℃(3 ℃/min),100 ℃保持1 min,100~285 ℃(4 ℃/min),285 ℃保持3 min;进样口温度,280 ℃;检测器温度,300 ℃;进样体积,1 μl;进样方式,不分流进样,尾吹气,30 ml/min。在该色谱条件下,对标准品峰的系统适应性参数进行考察,标准品峰理论塔板数均>4 000,分离度与对称因子符合中国药典要求[6]。供试品、对照品及叠加色谱图见图1~图3。

图1 1,3-二氯-2-丙醇与2,3-二氯-1-丙醇标准品色谱图

图2 供试品色谱图

图3 供试品与标准品色谱叠加图
2.6 方法学考察
2.6.1 线性范围与检测限 在5 支具塞试管中先分别加入0.5 ml 正己烷,再分别加入1 μl、3 μl、8 μl、15 μl、25 μl 标准使用溶液,加正己烷至1 ml,用气密针加入七氟丁酰基咪唑50 μl,立即塞紧,涡旋混合后,70 ℃保温20 min。放冷至室温,加饱和氯化钠溶液3 ml,涡旋混合30 s,使两相分离,取有机相加无水硫酸钠约为0.3 g 脱水,取溶液进行GC 测定。本方法检出限为0.01 mg/kg。线性关系见表1~表2。

表1 1,3-二氯-2-丙醇衍生化线性关系
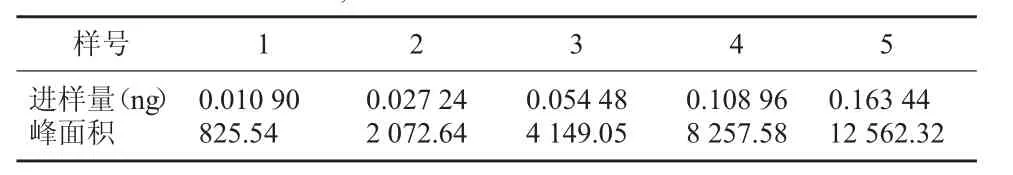
表2 2,3-二氯-1-丙醇衍生化线性关系
2.6.2 精密度试验结果 精密量取混合标准溶液200 μl,同2.3 法衍生后,进样6 针,精密度良好。见表3。
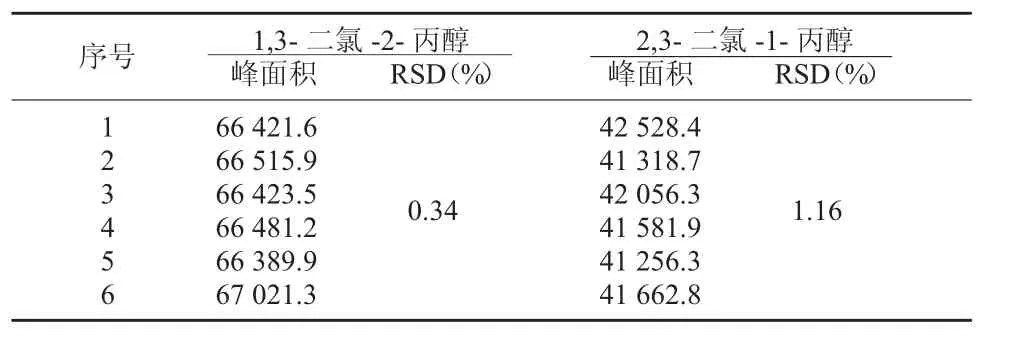
表3 精密度试验结果
2.7 回收率试验结果 精密取样品5 ml 中加入适量氯丙醇,同2.2 法净化和2.3 法衍生,进行回收试验,结果1,3-DCP 的加样回收率可达93.24%~96.41%,2,3-DCP 的加样回收率可达93.91%~96.51%。见表4。
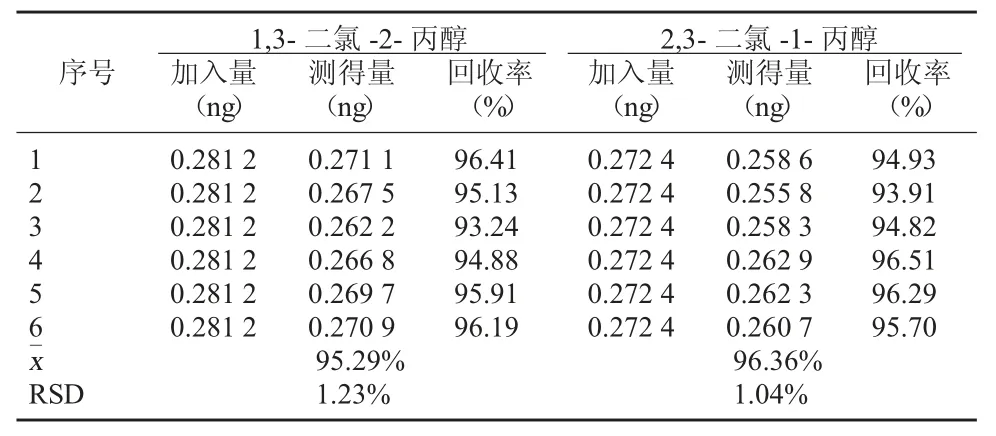
表4 回收率试验结果
2.8 6 批样品含量测定 取本公司初元复合肽营养饮品6 批,依照对样品中氯丙醇进行含量测定。见表5。
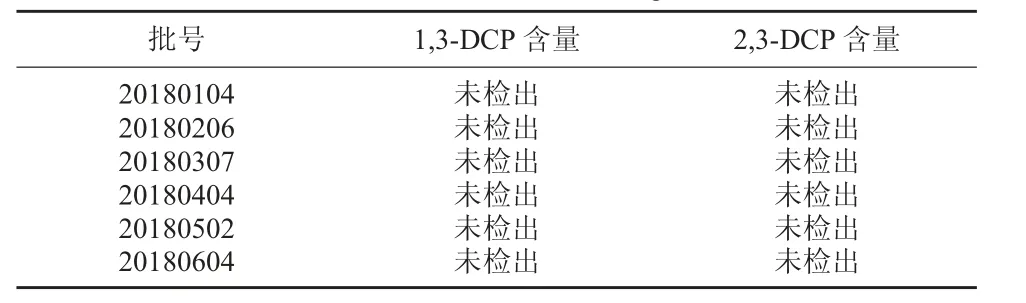
表5 6 批样品含量测定(ng)
3 讨论
通过多次重复性试验,发现样品经七氟丁酰基咪唑衍生化后得到的衍生物在非极性柱或弱极性柱的HP-5 毛细管柱进行分离,具有峰形好、出峰快的优势。试验中采用程序升温方式,在保证杂质峰与主峰能够高效分离的同时,也适当控制了各峰的保留时间。
当前随着人们物质生活水平的提高,食品安全问题受到社会越来越多的关注,对于有毒物质建立有效监控的检测方法成为食品安全生产中的重要一环。本试验结果显示,1,3-二氯-2-丙醇精密度为0.34%,回收率为95.29%,进样量为0.011 25~0.168 72 ng 时,峰面积与浓度呈良好线性关系(相关系数γ=0.999 9);2,3-二氯-1-丙醇精密度为1.16%,回收率为93.36%,进样量为0.010 90~0.163 44 ng 时,峰面积与浓度呈良好线性关系(相关系数γ=0.999 9)。方法学验证试验结果良好,所建立的方法操作简单、快捷,灵敏度高,试验结果准确可靠,适用于气相色谱法测定初元口服液产品的氯丙醇含量,也适用于生产企业进行质量控制。
