以肺部多发磨玻璃样结节影为主要表现的肺原发弥漫大B细胞淋巴瘤1例报告及文献复习
2020-06-12王荣荣
王荣荣,王 琦,李 伟,张 捷
(吉林大学第二医院呼吸内科, 吉林 长春 130041)
肺原发淋巴瘤(primary pulmonary lymphoma,PPL)是原发于肺内淋巴组织的恶性淋巴瘤,是结外淋巴瘤的一种罕见类型,多数为非霍奇金淋巴瘤(non-Hodgkin lymphoma,NHL),多起源于支气管黏膜相关淋巴结组织(mucosa-associated lymphoid tissue,MALT)。肺原发弥漫大B细胞淋巴瘤(diffused large B-cell lymphoma,DLBCL)是PPL的一种类型,该类型更为罕见,占PPL的10%~20%,占所有类型淋巴瘤的0.4%[1-2]。国内外研究[3-7]显示:肺原发DLBCL患者的胸部CT表现以单侧肺部团块影或实变影为主,患者中有多发性肺结节伴有胸腔积液和早期表现为渗出性病变者,也有空洞形成者。目前关于肺原发DLBCL患者影像学表现(多发磨玻璃样结节影)的文献报道较少。肺原发DLBCL患者因缺乏特异性临床表现,临床上容易漏诊和误诊。本文作者总结并分析本科诊治的1例以肺部多发磨玻璃样结节影为主要表现的肺原发DLBCL患者的临床资料,以提高临床医生对肺原发DLBCL的认识。
1 临床资料
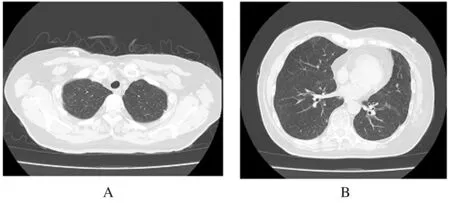
1.1 一般资料患者,女性,60岁,因“咳嗽和咳痰2个月”于2018年12月3日收入本科。该患者于入院前2个月无明显诱因出现咳嗽和咳痰。行胸部CT检查未见明显异常,自行口服莫西沙星后自觉症状好转,1个月前再次出现咳嗽和咳痰症状,于当地医院行肺部CT检查显示“多发结节影”,诊断为“肺炎”,行抗感染(莫西沙星联合哌拉西林舒巴坦)治疗2周后患者自觉症状好转,再次复查胸部CT(2018年12月3日):两肺纹理增多,两肺多发结节状磨玻璃密度影(图1)。患者为求进一步诊治就诊于本科。既往:体健。否认吸烟和大量饮酒史。查体:全身浅表淋巴结未触及肿大,听诊双肺呼吸音清,未闻及干、湿啰音和胸膜摩擦音。入院后患者行血常规、血沉、结核抗体、风湿系列相关检查、肿瘤标记物和痰液检查均未见明显异常。腹部彩超、腋窝和乳房彩超未见淋巴结异常。PET-CT检查提示:双肺多发磨玻璃结节伴部分代谢增高。结合患者的临床表现和辅助检查,建议患者进一步行纤维支气管镜取活检行组织病理学检查。
1.2 病理资料病理结果为(右肺上叶)NHL,大B细胞(图2,见封三)。免疫组织化学CD20(+),BCL-2(+),CD10(-),BCL-6(+/-),MUM-1(+),CD30(+),CyclinD1(-),C-myc约(30%+),Ki-67增殖指数大于50%,EBER(-)。最终诊断为肺原发DLBCL,病理诊断:非霍奇金DLBCL,起源于生发中心外活化B细胞。
1.3 治疗方法因患者符合DLBCL 诊断标准,最终诊断为肺原发DLBCL,故转入本院肿瘤科,给予R-CHOP方案进行化疗,按疗程规律治疗,随访3个月后再次复查胸部CT(2019年3月8日),检查结果显示:两肺纹理增强、紊乱和模糊,两肺多发类圆形结节状病变消失(图3)。

A:Superior lobe; B: Inferior lobe.
Fig.1 CT images of lung tissue of one patient with primary DLBCL at admission
A:Superior lobe; B: Inferior lobe.
Fig.3 CT images of lung tissue of one patient with primary DLBCL after regular treatment for 3 months
2 讨 论
PPL是一种较为罕见的结外淋巴瘤,定义为肺实质或支气管淋巴组织的克隆性异常增生,伴或不伴有肺门淋巴结肿大,在发病或确诊后3个月内无胸外淋巴瘤征象[8]。该患者全身淋巴结彩超未见异常淋巴结增大,骨髓穿刺未见恶性增殖淋巴细胞,随访3个月后未发现胸外淋巴瘤征象,病理免疫组织化学检测结果显示病理类型为DLBCL,诊断为肺原发DLBCL。
研究[9]显示:肺原发DLBCL是PPL的 第二常见病理类型,单纯 DLBCL 的起源尚无法明确,部分可由黏膜相关淋巴组织(MALT)淋巴瘤转化而来,故其临床和影像学特征与 MALT 淋巴瘤较为相似。肺原发DLBCL多见于中老年人和免疫抑制者,男女比例无明显差异[10],患者呼吸系统症状多表现为咳嗽、呼吸困难和胸痛,偶有咯血,全身症状包括发热、盗汗及体质量减轻等非特异性临床特征[11-12], 少数患者无临床症状,仅体检时发现肺部结节或阴影[13],诊断主要依赖组织病理学和免疫组织化学检查。
有研究[14]显示:PPL患者CT检查表现为单发或多发结节、团块或实变影,有时可见空气支气管征、病变周围晕征和血管造影征。而肺原发性DLBCL病变多发于下叶外周,CT检查多表现为边缘清晰的孤立多发结节影和团块影,也可表现为斑片状或不规则实变,伴或不伴磨玻璃样影,另外间质受累,病变浸润胸膜出现胸腔积液者亦有报道[15-16]。纵隔淋巴结节增大及胸腔积液较低度恶性淋巴瘤更为常见[17], 多层螺旋CT检查可表现为区域坏死性空洞[18],该例患者以胸部多发磨玻璃结节影为主要表现。
镜下DLBCL由弥漫成片的、大的母细胞性淋巴样细胞组成,可融合成群,破坏正常的肺组织,50%患者可出现局部淋巴结受累[11],研究[11-12]显示:肺原发DLBCL 患者的典型免疫组织化学表现为 CD20 和 CD79a 阳性及轻链限制, Ki67 增殖指数呈典型性升高。DLBCL是PPL的第二常见病理类型(约占PPL的10%),较MALT淋巴瘤(占PPL的70%~90%)更具侵袭性且预后差。两者治疗方案相似,但由于DLBCL局部或远处转移复发率较高,即使病变局限者手术切除后仍需要联合放化疗,高度恶性的大 B 细胞淋巴瘤一般不推荐手术,以化疗为主[19]。Ki67对预测DLBCL具有十分重要的意义,增殖指数大于60%提示预后较差。有研究[20]表明:DLBCL患者中约有45%出现B淋巴细胞瘤2基因(B-cell lymphoma-2,Bcl-2) 蛋白高表达,而高表达 BCL-2或骨髓细胞瘤原癌基因(myelocytomatosis oncogene, MYC)均会导致患者预后不良。另有研究[21]显示:所有DLBCL 患者中,并发 c-MYC 基因异常者约占 10%,且该类患者肿瘤细胞较 c-MYC 基因突变阴性者具有更强的增殖活性,提示 BCL-2 或 MYC和c-MYC 基因突变是 DLBCL 患者预后评估的重要指标[22-23]。该患者经规律化疗,3个月后复查胸部CT检查可见双肺磨玻璃影消失,结合基因检测标记,提示患者预后较好。
综上所述,肺原发DLBCL临床少见,当临床症状较轻,影像学却出现单发或多发的结节、团块或实变影或伴有空气支气管征、病变周围晕征和血管造影征,痰涂片及痰脱落等细胞学检查多为阴性,前期抗炎治疗效果不佳,病情进展或反复时,应考虑该病的可能性。本文作者分析1例以肺部多发磨玻璃样结节为主要表现的肺原发DLBCL患者的诊治经过,以提高临床医生对该类疾病的认识,减少误诊及漏诊。
