奥美沙坦酯的合成工艺优化
2020-06-10陈炜,杨健
陈 炜, 杨 健
(浙江大学 化学工程与生物工程学院,浙江 杭州 310027)
1 前 言
奥美沙坦酯 (4-(1-羟基-1-甲基乙基)-2-丙基-1-{4-[2-(四唑-5-基)苯基]苯基}甲基咪唑-5-羧酸(5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲基酯) 是由日本SankyoPharma研制的一种抗高血压药物,属于血管紧张素Ⅱ类受体拮抗剂[1],2002年5月经美国FDA批准上市,具有降压作用明确、药效稳定持久、副作用较小、耐受性好等优点[2]。奥美沙坦酯的专利保护期已过,但国内仿制药少,价格高,对其合成工艺的改进具有重要意义。
目前文献报道的奥美沙坦酯合成工艺,主要可以分为两类。一类是咪唑类中间体先与联苯类中间体烷基化、水解,再与4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮酯化,经过脱保护得到奥美沙坦酯[3-9];第二类是咪唑类中间体先与4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮酯化,再与联苯类中间体经过烷基化反应、水解等步骤得到奥美沙坦酯成品[10]。不同路线的区别主要有两点,一是关键中间体的结构不同,二是合成过程采用的催化剂种类不同。文献报道大多采用格氏试剂、氢化钠或者叔丁醇钾等作为催化剂,存在催化剂价格昂贵、反应条件苛刻、反应不易控制等问题,且得到的中间体杂质较多,需要引入额外的纯化步骤,不利于工业化生产。收率方面,文献报道总收率范围为45%~55%,存在较大提升空间。
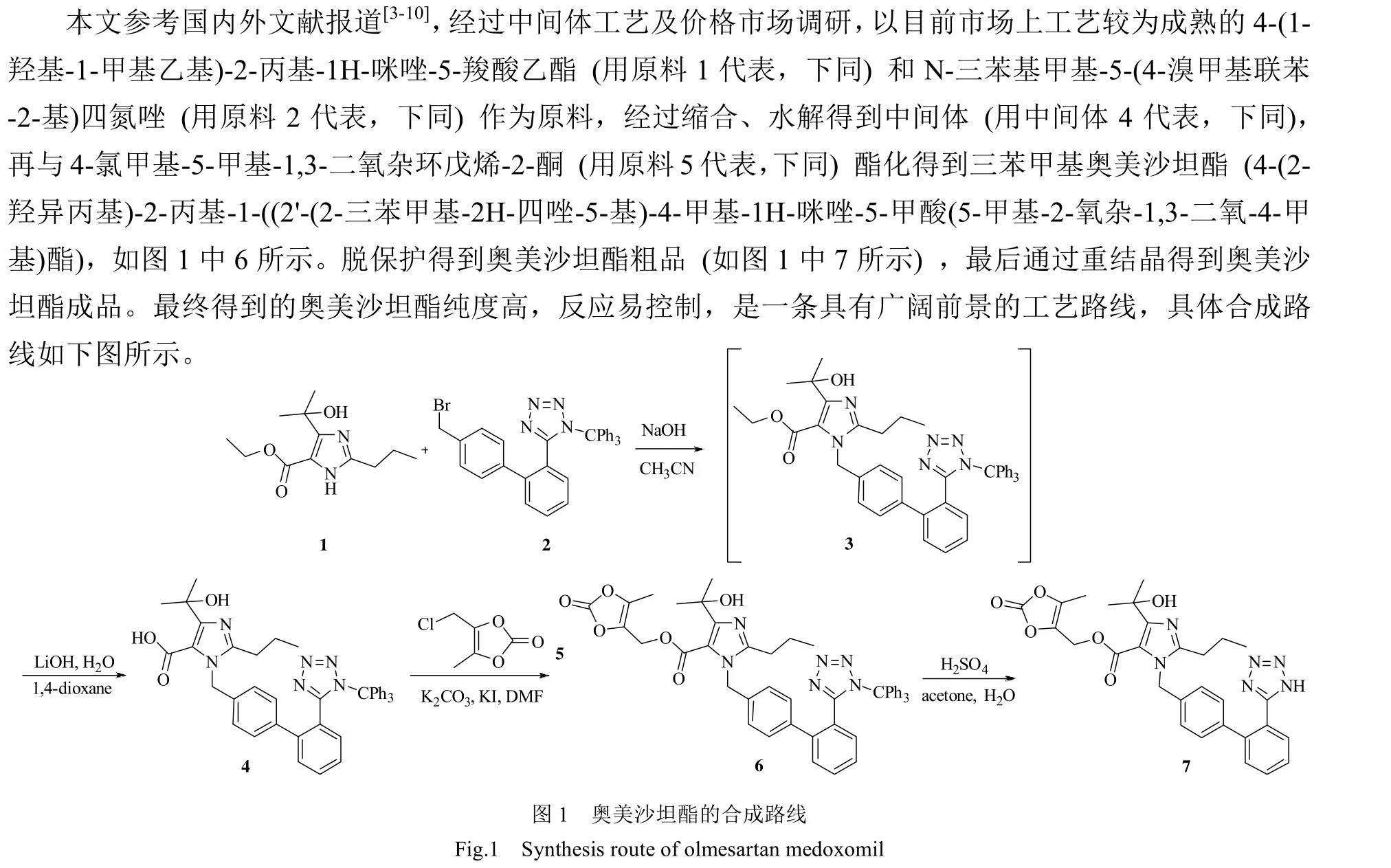
2 实验部分
2.1 分析仪器及试剂
WRS型熔点仪,Bruker Esquire-LC质谱(MS)分析仪,Bruker Avance DMX 500 核磁共振仪,Shimadzu LC-20A高效液相色谱 (HPLC) 仪。乙腈等其他试剂均为分析纯。
2.2 奥美沙坦酯的合成
2.2.1 中间体4的合成
称取30.0 g 原料1、76.7 g 原料2、7.5 g的氢氧化钠和790.0 g溶剂乙腈,加入至2 L的四口反应瓶中,机械搅拌,此时体系呈固液两态。水浴加热,体系升温至70 ℃保温反应。随温度升高,体系逐渐溶清。薄层色谱法 (thin layer chromatography, 简称TLC) 检测反应进程,待反应基本完全,则停止反应。体系稍降温后,40 ℃下减压蒸除大部分溶剂至无明显液滴流下,体系呈棕褐色黏稠液体。向其中加入水解溶剂1,4-二氧六环共300.0 g,并转移至2 L的四口反应瓶中,待用。
将体系加热至40 ℃,机械搅拌。称取18.2 g一水合氢氧化锂溶于100.0 g水中,缓慢滴加到体系中,保温使其继续反应。TLC检测,待反应基本完全,则停止加热,加入乙酸乙酯320.0 g和饮用水300.0 g萃取产物。继续搅拌,缓慢滴加75%的冰乙酸水溶液调节体系pH至6-7,过程中有大量白色固体析出。将体系降温至10 ℃左右继续搅拌,使产物充分析出。抽滤,滤饼用乙腈打浆0.5 h。再次抽滤,滤饼40 ℃减压干燥,得中间体4 (4-(1-羟基-1-甲基乙基)-2-丙基-1-[[2'-[三苯甲基-1H-四唑-基][1,1'-联苯]-4-基]甲基]-1H-咪唑-5-羧酸) 73.4 g,经HPLC测定质量百分数为98.5%。
HPLC条件:色谱柱为C18,波长为230 nm,流动相为乙腈:水(pH 3.0)=80:20(V/V),流速为1.0 mL⋅min-1,运行时间为30 min。
中间体4结构表征结果如下:
1H-NMR(DMSO-d6):δ(10-6) = 0.72~0.77 (3H,t,CH3),1.46~1.54 (2H,q,CH2),1.57 (6H,s,2 CH3),2.41~2.50 (2H,t,CH2),5.61 (2H,s,CH2),6.88~6.90 (8H,d,苯环H),7.03~7.06 (2H,d,苯环H),7.30~7.37 (9H,q,苯环H),7.42~7.45 (1H,q,苯环H),7.49~7.61 (2H,m,苯环H),7.77~7.80 (1H,d,苯环H)。
2.2.2 三苯甲基奥美沙坦酯的合成
称取60.0 g中间体4、24.1 g碳酸钾、330 g溶剂N,N-二甲基甲酰胺(N,N-dimethylformamide,简称DMF),加入至2 L四口反应瓶中,25 ℃下机械搅拌1 h,体系逐渐溶清。向其中加入7.2 g的碘化钾,并缓慢滴加原料5共18.1 g,室温 (25 ℃) 下继续反应。TLC检测,待反应基本完全,则停止反应。将体系抽滤,向滤液中加入乙酸乙酯350.0 g、饱和食盐水450.0 g进行萃取,静置分层,水层用120.0 g乙酸乙酯继续萃取三次,合并有机层,有机层经过饱和食盐水洗涤、无水硫酸钠干燥后,40 ℃减压蒸除大部分溶剂至无明显液滴流下,向其中加入乙腈209 g,10 ℃搅拌1 h,有大量白色固体析出,抽滤,滤饼40 ℃减压干燥,得到三苯甲基奥美沙坦酯(中间体6)63.1 g,经HPLC测定质量百分数为99.4%。
HPLC条件同中间体4的条件。
中间体6结构表征结果如下:
1H-NMR(CDCl3):δ(10-6) = 0.88~0.92 (3H,t,CH3),1.62 (6H,s,2 CH3),1.66~1.68 (2H,q,CH2),1.98 (3H,s,CH3),1.99 (1H,s,N结合水峰),2.52~2.56 (2H,t,CH3),4.76 (2H,s,CH2),5.30 (2H,s,CH2),5.589 (1H,s,OH),6.68~6.70 (2H,d,苯环H),6.95~7.00 (6H,d,苯环H),7.08~7.10 (2H,d,苯环H),7.25~7.29 (7H,q,苯环H加氘代氯仿出峰),7.32~7.36 (3H,m,苯环H),7.40~7.51 (3H,m,苯环H),7.86~7.88 (1H,d,苯环H)。
2.2.3 奥美沙坦酯粗品的合成
称取50.0 g的中间体6、80.0 g丙酮、100.0 g纯化水,加入至2 L四口反应瓶中,机械搅拌,此时体系呈固液两态。25 ℃下缓慢滴加浓硫酸14.7 g,体系逐渐溶清。搅拌继续反应,约0.5 h后逐渐有白色固体析出。TLC检测,待反应基本完全,则停止反应。抽滤,滤去生成的白色固体,反应瓶以及滤饼用160.0 g纯化水润洗,收集滤液并降温至5 ℃左右,继续搅拌0.5 h后抽滤,滤去析出的少量白色固体,此时滤液澄清透明。缓慢滴加30%的碳酸钾水溶液调节pH至6~7,体系逐渐浑浊,有白色固体析出,继续搅拌0.5 h,抽滤,滤饼40 ℃降压干燥。此时所得体系中含有大量无机盐硫酸钾,以纯化水水洗除去无机盐,抽滤,滤饼40 ℃减压干燥,得到奥美沙坦酯粗品31.2 g,经HPLC测定质量百分数为99.5%。
HPLC条件:色谱柱为C8,波长为256 nm,流动相A为配置2.04 g⋅L-1的磷酸二氢钾溶液,以1.73 g⋅L-1的磷酸溶液调节pH至3.4,将此溶液以体积比4:1与乙腈混合,流动相B为配置2.04 g⋅L-1的磷酸二氢钾溶液,以1.73 g⋅L-1的磷酸溶液调节pH至3.4,将此溶液以体积比1: 4与乙腈混合,流速为1.0 mL⋅min-1,柱温为40℃[11]。
2.2.4 奥美沙坦酯的合成
称取30.0 g奥美沙坦酯粗品、270.0 g丙酮和30.0 g纯化水,加入到500 mL的四口反应瓶中,升温至40 ℃使其溶清,趁热过滤除去其中机械杂质。体系35 ℃减压蒸除部分溶剂至刚好出现浑浊,搅拌并加入120.0 g乙酸乙酯,使产品充分析出,得到奥美沙坦酯成品 (27.0 g,HPLC:99.8%)。
HPLC分析条件同中间体6的条件。
产品结构表征结果如下:
熔点(melting point):177~178℃
MS (ES-) 557.2,MS (ES+) 559.3
1H-NMR(DMSO-d6):δ (10-6) = 0.90~0.93 (3H,t,CH3),1.52 (6H,s,2 CH3),1.60~1.65 (2H,q,CH2),2.11 (3H,s,CH3),2.63~2.66 (2H,t,CH2),5.10 (2H,s,CH2),5.22 (1H,Brs,OH),5.46 (2H,s,CH2),6.90~6.91 (2H,d,苯环H),7.08~7.104 (2H,d,苯环H),7.56~7.58 (2H,m,苯环H),7.59~7.61 (2H,m,苯环H)。
3 结果与讨论
3.1 中间体4的合成工艺条件的选择及优化
中间体4的合成反应为强碱作用下的缩合反应,遵循单分子亲核取代反应(SN1)机理,文献报道多采用氰化钠或叔丁醇钾作为催化剂,在溶剂DMF中发生反应,催化剂价格昂贵且反应收率低。而乙腈对于奥美沙坦酯及其中间体是一种良好的结晶溶剂,以乙腈为溶剂进行缩合反应所得的中间体中杂质明显少于DMF体系[6]。采用氢氧化钠代替昂贵的氰化钠或者叔丁醇钾,副反应杂质大大减少,且反应温和可控。此体系副反应的发生与碱的用量、反应温度等有关。添加碱的量过多、反应温度过高会导致副产物的增加,从而造成中间体纯度的下降。控制以上参数可以使副反应降低,再经过水解反应的后处理步骤可得到纯度较高的中间体4。
首先考察原料1和原料2的投料比对反应的影响。以更为廉价的原料1为1.0当量(equivalent,eq),考察结果显示,在其他反应条件相同的情况下,原料2用量为0.9eq和1.0eq时,原料1均有较多的剩余。原料2用量为1.1eq以上时可以使原料1基本反应完全,而原料2用量为1.2eq以上时导致原料2过量,因此原料1和2的投料比以1.1eq为佳。具体实验结果如表1所示。

表1 原料2的当量对反应的影响 Table 1 Effects of raw material 2 equivalent on reaction
其次,考察碱的用量对反应的影响,实验结果列于表2。可以看出,NaOH的用量在1.4~1.6eq时,原料剩余较少,反应基本进行完全,因此最佳的NaOH用量可确定为1.5eq。随着碱用量的进一步增加,收率有所下降,可能是因为副反应增加所致。再次,考察温度对反应的影响,实验结果列于表3。可以看出,反应温度低于55 ℃时,反应速率相对较慢,延长反应时间考察,原料仍有较多剩余;反应温度范围为65~75 ℃时,原料剩余较少,反应基本进行完全;温度继续升高达到回流温度82 ℃时,副反应的增加导致产品收率下降。

表2 NaOH当量对反应的影响 Table 2 Effects of NaOH equivalent on reaction
在缩合步骤结束后单独分离出中间体,需花费额外的后处理以及烘干储存时间,从而造成成本的提升。经实验考察,缩合反应得到的产物中含有部分杂质,杂质可在水解反应的后处理步骤中被除去,因此中间体可不单独分离,而是蒸除部分溶剂后加入水解溶剂和碱直接进行水解反应。将缩合反应和水解反应合并的操作,还有效的减少了分离缩合反应产物过程中杂质的产生,提高了反应收率。

表3 温度对反应的影响 Table 3 Effects of temperature on reaction
在水解反应所使用的溶剂方面,对中间体4溶解性较好的常用溶剂有1,4-二氧六环和异丙醇,在40 ℃下分别进行水解反应4 h,结果如下:以1,4-二氧六环为溶剂的得到产物纯度在97%左右,而以异丙醇为溶剂得到的产物纯度为83%和93%,产品质量不高且波动较大,因此确定以1,4-二氧六环为溶剂较佳。虽然1,4-二氧六环相对毒性较大,药典规定在成品中的残留需小于380×10-6,而反应后续还有酯化、脱保护以及重结晶等多个步骤,残留的1,4-二氧六环在其中的含量不断减少,通过气相色谱法(GC)对最终成品进行检测,色谱柱型号为DB-624,载气为氮气,载气流速为4.6 mL⋅min-1,分流比为5:1,氢气流速为50 mL⋅min-1,空气流速为450 mL⋅min-1,检测器类型为火焰离子化检测仪(flame ionization detector, 简称FID)[11]。结果表明,1,4-二氧六环的浓度低于检测限5.5×10-6。
考察碱的种类对水解反应的影响。40 ℃下水解4 h,发现氢氧化钠体系得到的产物杂质较多,收率在80%左右;采用相同当量碱性相对较弱的氢氧化锂,收率可达到95%,因此选择氢氧化锂作为水解碱。 考察氢氧化锂当量对水解反应的影响,以缩合反应中的原料1的量为1.0eq,实验结果列于表4。可以看出,氢氧化锂为2.0eq下进行水解反应,中间体3有较多剩余。氢氧化锂为3.0和4.0eq下,中间体3均基本反应完全,且收率较高,因此,水解反应时取氢氧化锂当量3.0eq为佳。
在反应时间方面,从表5可以看出,反应在3 h左右已基本进行完全,再延长反应时间,副反应继续发生导致收率有所下降。
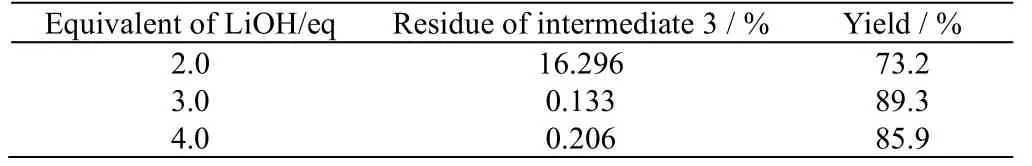
表4 LiOH当量对反应的影响 Table 4 Effects of LiOH equivalent on reaction

表5 时间对反应的影响 Table 5 Effects of reaction time on reaction

表6 原料5的当量对反应的影响 Table 6 Effects of raw material 5 equivalent on reaction

表7 时间对反应的影响 Table 7 Effects of reaction time on reaction
3.2 三苯甲基奥美沙坦酯的合成工艺条件选择及优化
三苯甲基奥美沙坦酯的合成反应为酯化反应,是羧酸根负离子对卤代烃α碳的亲核取代反应[12],所采用的原料5因具有基因毒性,需要严格控制其加入量[13],通过缓慢滴加来控制其用量。另外,考虑到原料5的活性较低,反应速率较慢,预先加入适量碘化钾将滴加的原料5转化为具有较高反应活性的碘代物,可与已在碱性环境下转化为相应盐的中间体4快速进行反应,从而减少原料5的分解。得到的中间体经过乙腈、甲醇后处理可除去生成的少量副反应杂质,得到纯度较高的三苯甲基奥美沙坦酯(中间体6)。 首先考察加入的原料5量对反应的影响,中间体4的量为1.0eq时,实验结果列于表6。可以看出,原料5的用量在1.2eq以上时可使反应基本进行完全。考虑到产品中残留的中间体4会随着后续脱保护反应的进行转化为欧洲药典(EP)所规定的杂质A,难以除去,因此中间体4在产物当中的含量需要进行严格控制,原料5的用量以1.2eq为佳。
考察碳酸钾用量对反应的影响,碳酸钾用量1.0、2.0以及3.0eq时均可使反应进行完全,且对产品质量无明显影响,因此反应取1.0eq的碳酸钾即可。
在反应时间方面,从表7可以看出,添加了碘化钾作为活化剂后,反应在室温下即可快速进行,1 h左右已基本反应完全,随着反应时间进一步延长,可能导致副产物的增加,产品收率下降。
3.3 奥美沙坦酯粗品的合成条件的选择及优化
关于中间体6中的三苯甲基的脱除反应,文献报道较多。据早期文献报道,采用盐酸或醋酸作为酸催化剂、低温下脱保护[1]时,由于盐酸极易挥发,加入体系后部分挥发导致体系上方被白雾笼罩,不易观察体系变化,且盐酸或醋酸下脱保护不完全,存在较多中间体6的残留,进而导致奥美沙坦酯粗品纯度较低。若延长反应时间则又会导致奥美沙坦酯在酸性环境下进一步水解生成奥美沙坦这一杂质,难以除去,需经过数次重结晶才能达到EP标准。SRIMURUGAN等[14]报道,在强碱条件下醇溶液中可脱除氯沙坦钾中的三苯甲基保护基团,但此方法缺点是副反应较多,所得产品中含有较多杂质。HEDVATI等[15]提出,以乙腈和水的混合液溶解三苯甲基奥美沙坦酯,直接加热回流条件下可以脱除三苯甲基,也有利用丙酮[16]、乙醇[17]等与水的混合液回流进行脱保护的相关报道,但是这些方法缺点是回流温度较高、脱保护速率较慢、产品纯度不高。而采用不挥发性酸,如浓硫酸进行脱保护,实验结果表明,中间体6几乎反应完全,而反应产生的三苯基甲醇不溶于体系,可直接过滤除去,再通过调节pH使奥美沙坦酯粗品析出。硫酸加入体系时会放热,因此反应需严格控制浓硫酸滴加速率、体系搅拌速率以及体系温度。通过工艺参数的优化,最终的脱保护工艺条件下得到的奥美沙坦酯粗品纯度高,粗品即可符合EP标准[11],再经过进一步重结晶可得到更优的奥美沙坦酯成品。
考察浓硫酸用量对反应的影响,以中间体6的用量为1.0eq,实验结果列于表8,可以看出,浓硫酸的量在2.0eq以上时,反应基本进行完全,考虑到过量的浓硫酸可能导致放热效应增加,且后处理需要更多的碱性溶液进行中和析晶,从而产生大量废水,因此,浓硫酸当量取2.0eq为最佳。
在反应过程中,温度可能对杂质的产生有影响,实验结果列于表9,可见,随着温度升高,EP记载的杂质A(olmesartan)的含量明显增加,杂质C ((5methyl2oxo1,3dioxol4yl)methyl 4(1methylethenyl) 2propyl1[[2′(1Htetrazol5yl)biphenyl4yl]methyl]1Himidazole5carboxylate)的含量在此温度范围内没有明显差异,杂质B(6,6dimethyl2propyl3 [[2′(1Htetrazol5yl) biphenyl4yl]methyl] 3,6dihydro4Hfuro [3,4d]imidazol4one)在此过程中未检测到。因此,在实际生产中应严格控制反应温度,取15~25 ℃即可。
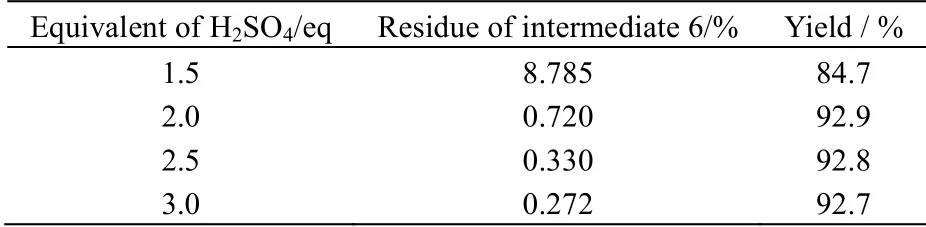
表8 浓硫酸的量对反应的影响 Table 8 Effects of H2SO4 equivalent on reaction

表9 温度对反应的影响 Table 9 Effects of temperature on reaction
3.4 重结晶条件的选择及优化
丙酮与水的混合溶液对奥美沙坦酯的溶解性较好,因此,目前文献报道往往采用丙酮作为奥美沙坦酯的重结晶溶剂。但是此方法存在一个严重的问题,即丙酮体系的析晶产物易形成溶剂化物[18],丙酮-水溶液直接析晶的成品中,丙酮的残留在9000×10-6以上,不符合药典规定的溶剂残留标准(5000×10-6)。孙宝佳等[19]采用粉碎再烘干法对其中的残留丙酮进行脱除,额外粉碎、烘干步骤导致能耗增加、成本升高。经实验考察,在丙酮析晶过程中加入适量乙酸乙酯进行搅拌析晶,烘干至衡重后用GC检测最终成品中的乙酸乙酯和丙酮的残留均符合药典规定,丙酮残留最低至500×10-6左右,此方法具有操作简单,成本低的优势。
考察析晶过程中加入的乙酸乙酯量对反应的影响,以奥美沙坦酯的量为1.0eq,析晶时间2.0h,实验结果列于表10,可以看出,加入4.0eq的乙酸乙酯时,重结晶产品中两种溶剂的残留均较少,同时收率也较高。乙酸乙酯加入量较少时,丙酮残留相对较多;乙酸乙酯加入量较多时,产品中残留的乙酸乙酯增加,且部分产品溶解于乙酸乙酯中,导致收率下降。在4.0eq的乙酸乙酯条件下,进一步延长析晶时间到4.0 h,重复三次实验收率分别为87%、85%和90%。综合整个反应,总收率达到65.5%。

表10 乙酸乙酯当量对反应的影响 Table 10 Effects of ethyl acetate equivalent on reaction
4 结 论
(1) 以4-(1-羟基-1-甲基乙基)-2-丙基-1H-咪唑-5-羧酸乙酯和N-三苯基甲基-5-(4-溴甲基联苯-2-基)四氮唑为原料,经缩合、水解、酯化、脱保护,四步反应合成了奥美沙坦酯。合成总收率达到65.5%,较文献值提高10%以上,该路线操作简单、反应温和、适宜工业化生产。
(2) 缩合与水解反应合并,减少了中间体分离烘干工序;采用温和的无机碱催化反应,两步总收率达到89.3%。以碘化钾活化4-氯甲基-5-甲基-1,3-二氧杂环戊烯-2-酮进行酯化反应,反应1 h即可进行完全,收率达到92.2%。以浓硫酸为酸催化剂进行脱保护反应,反应进行的彻底、中间体残留少,奥美沙坦酯粗品纯度即可基本满足欧洲药典标准,收率达到93.7%。
(3) 重结晶过程中加入适量乙酸乙酯,可有效地减少产品中丙酮的残留,达到药典标准。
