微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓
2020-06-09张荣谢有超喻昌木彭黔荣杨敏
张荣,谢有超,喻昌木,彭黔荣,,杨敏,3
(1 贵州大学化学与化工学院,贵州贵阳550025;2 贵州中烟工业有限责任公司技术中心,贵州贵阳550009;3 贵州大学药学院,贵州贵阳550025)
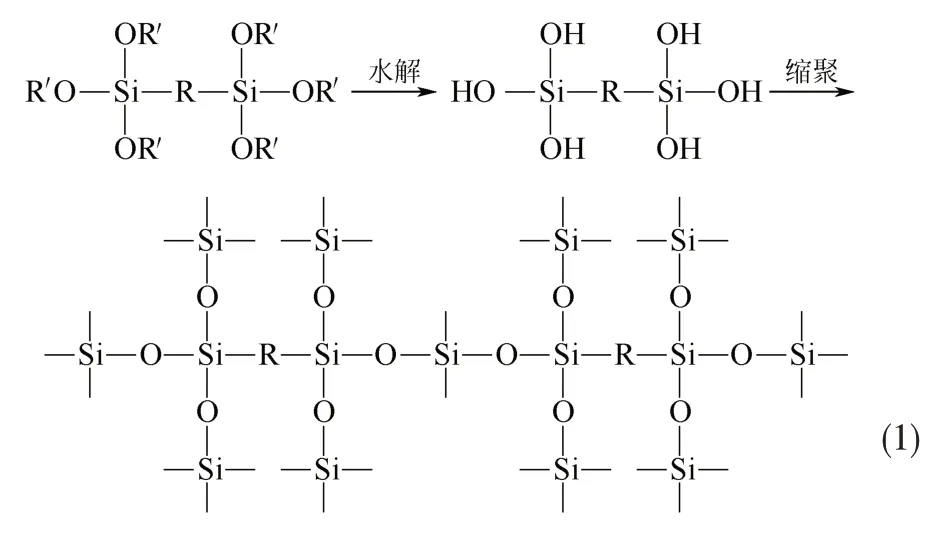
有序介孔有机硅材料(PMOs)具有较高的比表面积,孔隙结构发达,且有着良好的热化学稳定性和机械稳定性[1-2],所以常被用于气体吸附[3]、分离[4]和催化领域[5]。通常PMOs 的合成方法如图1 所示。双硅氧烷化的有机硅前体在酸或碱的水溶液中自身水解缩合,桥联倍半硅氧烷单体水解缩合反应见式(1),经过去除模板剂得到有序介孔有机硅材料[6-7]。但是,普通双硅氧烷化的有机硅前体活性位点较少,对其水解产物进行后合成修饰条件苛刻,且容易造成材料的垮塌[8]。而双硅氧烷化的1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓除自身的活性位点以外,还具有可供修饰的阴离子,通常通过阴离子交换反应可以将阴离子类型的功能基团修饰到1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓上[9-12],将修饰后的1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓固载化成为功能化有序介孔有机硅材料(PMO-IL)(见表1)。
目前,文献中常见的合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的方法有甲苯为溶剂[13]、乙腈为溶剂[14]和无溶剂[15]3 种方法;但这些方法都存在耗时长、产率低、且需要消耗大量有机溶剂和冷凝水等缺陷,不符合绿色化学的要求。微波辐照合成法因为加热速度快、物料受热均匀和特殊的非热效应被广泛用于有机合成反应中,且该法具有操作方便、产物收率高、绿色无污染等优点[16]。目前,采用微波辐照法合成单硅氧烷化的有机硅前体已经做了大量的研究工作[17],但将微波辐照法用于双硅氧烷化的1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的合成还未见报道。在此,本文首次将微波辐照法用于1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的合成,并比较了3 种常规方法和微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的收率情况。
1 实验部分
1.1 实验仪器与试剂
格兰仕家用微波炉[G80F23CSL-Q6H(B0)],广东格兰仕微波炉电器制造有限公司;傅里叶变换红外光谱仪(FTIR,TENSOR-27,KBr 压片),德国Bruker光谱公司;电子天平(ALC-110.4),德国艾科勒;旋转蒸发仪(RE52-99),上海亚荣生化仪器厂;循环水真空泵(SHZ-Ⅲ),浙江黄岩黎明实业有限公司;磁力搅拌器(85-1A),巩义市予华仪器有限责任公司;真空干燥箱(DZF-6020),郑州贝楷仪器有限公司;恒温鼓风干燥箱(DHG-9070),上海郎轩实验设备有限公司。
N-(3-乙氧基硅丙基)咪唑硅烷,实验前预先合成,并过柱子纯化;3-氯丙基三乙氧基硅烷(98%),上海麦克林生化科技有限公司;乙腈、乙醚、乙酸乙酯均为分析纯,天津市富宇精细化工有限公司。
1.2 合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓
常规方法和微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的反应机理一致,均为季铵化反应,如式(2)所示。
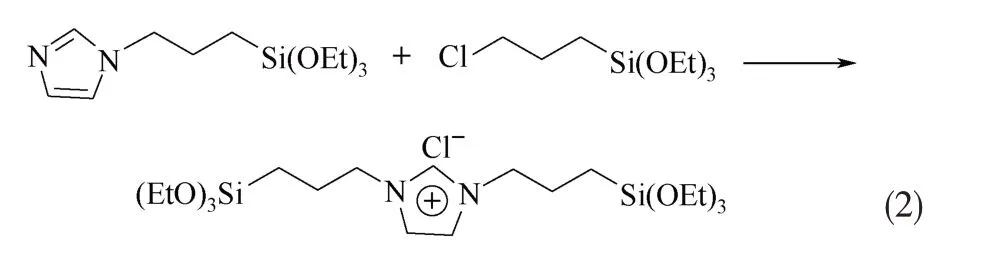
1.2.1 常规方法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓
准确秤取N-(3-乙氧基硅丙基)咪唑硅烷(3.7 mmol,1g)于装有搅拌子的25mL圆底烧瓶,加入一定量溶剂并搅拌使其充分混匀;将圆底烧瓶移到一定温度的油浴锅中,搅拌条件下缓慢滴加3-氯丙基三乙氧基硅烷(5.55mmol,1.5mL),加毕,充N2保护,TLC监控反应进程;反应过程中体系颜色逐渐从淡黄变为棕色,反应结束后用乙醚(5 次×10mL)充分洗涤棕色液体,移去洗液即得纯的目标产物,称重并计算产率。

图1 由桥联倍半硅氧烷单体合成PMOs示意图

表1 各种PMOs材料对应前体结构和功能化方式比较
1.2.2 微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓
准确秤取一定量的N-(3-乙氧基硅丙基)咪唑硅烷与3-氯丙基三乙氧基硅烷于25mL圆底烧瓶,摇匀;用N2置换圆底烧瓶内的空气,重复3次,然后在瓶口套1 个干净的气球。先设定一系列微波功率,确定不同微波功率条件下单次微波辐照时间;然后在确定的微波功率和单次微波时间条件下,研究投料比、微波辐照总时长、投料量对1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓收率的影响;产物后处理方法和常规合成方法完全一致。
2 结果与讨论
2.1 3 种常规合成法对1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓产率的影响
溶剂和温度条件对该反应的影响较大,目前的文献报道有用甲苯和乙腈作溶剂,也有不用溶剂的做法。在此,本文考察了以甲苯、乙腈为溶剂和无溶剂条件下3 种方法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓产率情况。
季铵化反应的机理是SN2亲核取代,其中溶剂和反应温度被认为是影响季铵化反应的两大主要因素。最初,Kondo 等[18]经过多次试验提出溶剂极性越大季铵化反应速率越快的论点;后来Guiuen等[19-20]在研究吡啶季铵化反应后认为溶剂介电常数越高,季铵化反应速度越快;刘劲松等[21]在连续型季按化反应的热动力学研究中发现,温度升高会增大季铵化反应速率常数。在此,本文对不同溶剂条件下合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的收率和产物外观特性进行了比较,结果如表2所示。

表2 3种常规合成方法对应产物收率
由表2可知,相同反应时间内无溶剂体系对应产品收率低于甲苯和乙腈溶剂体系(条目1,2 和3,4),这是因为无溶剂反应体系中反应物分子之间的传质速率较慢,减缓了反应进程。同时,溶剂的极性对该季铵化反应有很大影响,乙腈作溶剂时反应36h产品收率就能达到77%,而甲苯作溶剂时反应40h 产品收率仅68%,这是因为乙腈为极性溶剂,极性环境中3-氯丙基三乙氧基硅烷的碳卤键电子云分布发生改变,使碳原子的正电性增加,从而促进亲核取代反应的发生(条目1,3)。然而,无溶剂体系反应时间延长到45h后产品收率达81%,高于甲苯和乙腈溶剂体系。这可能是溶剂效应和温度效应共同作用的结果,即乙腈虽然会加快该季铵化反应的进行,但其反应温度较低限制了该反应的转化率;同时,甲苯溶剂体系虽然能达到较高的反应温度,但低极性的甲苯对该季铵化反应不利,只能加快反应到达平衡的速度,却限制了反应的转化率。由此可知,极性较大的溶剂和较高的反应温度对该季铵化反应有利。各反应体系中产品颜色不同是因为其浓度不同导致的,纯化后的1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓呈棕褐色。
综上可知,常规合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的方法反应时间太长,且需要消耗大量热量和冷凝水,不符合绿色化学要求。所以,有必要研究开发一种快速、高效合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的新方法。
2.2 微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓
微波辐照法不仅具有类似于传统热合成方法的热效应,还具有特殊的非热效应,而这种非热效应被认为是微波辐照合成法优于传统热合成法的主要原因。比如,Loupy[22-23]、Regina[24]等,均使用实验证明了微波作用于有机合成反应存在非热效应;Rosana等[25]分别利用常规加热和微波加热方法,在80℃的条件下反应250min来合成2-苄氧基-1-甲基吡啶鎓(BArF)。结果表明,微波法产物收率高达90%,而常规方法产物收率仅25%。这说明微波辐照用于有机反应具有加快反应速率、提高产物收率、减少反应所需能耗等优点。
在此,通过不断优化微波反应条件,使得产物氯鎓盐收率达到较高的水平。并且得出微波合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的最佳条件。与常规方法相比,微波法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓用时短、产率高、更符合绿色化学原则。对快速高效合成该类季铵盐具有十分重要的指导意义。
2.2.1 微波功率和单次微波时间确定
反应初期体系中原料极性较小,对微波能量的吸收较弱,温度升高较慢。随着微波时间的增加,生成极性较大的氯鎓盐快速吸收微波能量,使得体系温度迅速升高。当温度升高到一定值后,部分原料开始气化并产生白色烟雾,使反应不可控。所以,本文采用间歇微波辐照的方式对反应体系加热,即微波辐照一段时间后,将反应体系温度降低并搅拌均匀,再进行下次微波辐照,如此循环,直到反应结束。
在投料比N-(3-乙氧基硅丙基)咪唑硅烷∶3-氯丙基三乙氧基硅烷=1∶1(均为4mmol),且微波总时长为15min条件下,考察不同微波功率和对应单次微波时长对产品收率的影响,实验结果见表3。
选取各微波功率条件下体系沸腾的临界时间作为单次微波时长。由表3 可知,当微波功率为160W、240W、320W和400W时,对应单次微波时长分别为22s、15s、12s、10s,单次微波时长随微波功率逐渐减小。对比不同条件下产品收率可知,微波功率为240W 时对应产物收率最高为85.3%。当微波功率为160W 时,产物收率仅71.2%,这是由于较低的微波功率难以促使反应快速进行,使得原料在一定的反应时间内不能充分转化。当微波功率由240W升高到320W或400W时,产物收率略微下降,这是因为微波功率太大使得部分产物气化或分解。所以,最合适的微波功率为240W,单次微波时长15s。

表3 不同微波功率条件下单次微波时长和产物收率
2.2.2 微波辐照总时长对产物收率影响
微波功率240W,单次微波辐照时长15s 条件下,投料比N-(3-乙氧基硅丙基)咪唑硅烷∶3-氯丙基三乙氧基硅烷(CPTES)=1∶1(均为4mmol),调节微波辐照总时长,考察微波辐照时长对产品收率的影响,实验结果见表4。
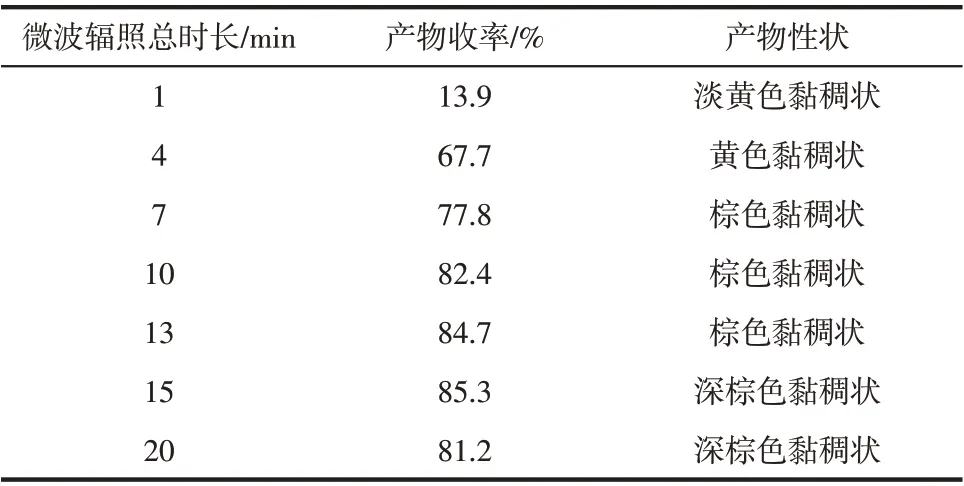
表4 微波辐照总时长对产物收率影响
由表4可知,微波辐照总时长1min时,收率仅13.9%,这是因为反应初期原料极性较小,吸收的微波能量较少,使得体系达不到反应所需温度,所以产率很低。而微波辐照总时长4min 时,产物收率突然增加到67.7%,这是因为随着微波时间增加,生成的季铵盐能够快速吸收微波能量,使体系温度快速升高,加快了季铵化反应的进程,所以产物收率迅速增加。微波总时长超过10min以后,产物收率增加不明显,一直到15min 达到最大收率,这是因为该阶段大部分反应物转化为季铵盐,剩余少量反应物之间传质受阻,导致反应速度减缓。当微波辐照总时长超过15min 以后,产物收率下降,颜色变深,这是因为体系升温过快使得部分产物分解。所以,最合适的微波辐照总时长为15min。
2.2.3 投料比对产物收率影响
在上述优化条件下,调节N-(3-乙氧基硅丙基)咪唑硅烷与3-氯丙基三乙氧基硅烷摩尔比(中间体为4mmol),考察投料比对产物收率的影响,实验结果见表5。

表5 投料比对产物收率影响
由表5可知,随着3-氯丙基三乙氧基硅烷投料量的增加,产物收率有上升的趋势。当3-氯丙基三乙氧基硅烷的投料量超过1.2n(中间体)时候,产物收率几乎不再增加,所以最合适的投料比为n(中间体)∶n(CPTES)=1∶1.2。
2.2.4 投料量对产物收率影响
微波功率240W,单次微波辐照时长15s,反应总时长15min,投料比n(中间体)∶n(CPTES)=1∶1.2条件下,改变N-(3-乙氧基硅丙基)咪唑硅烷的投料量,考察投料量对产物收率的影响,实验结果见表6。

表6 投料量对产物收率影响
由表6可知,N-(3-乙氧基硅丙基)咪唑硅烷投料量大于4mmol时,随着实验样品量增加产物收率略微减小,这是受实验设备的限制使体系中反应物之间不能充分接触造成的。而且,当实验样品量由4mmol减小到2mmol时产品收率降低了1.7%,这可能是由于反应提前完成,产物因过热而分解造成的。由此可知,一定反应条件下实验样品量增加对产品收率的影响很小,这得益于微波辐照合成法有着传统合成方法所不具备的非溶剂效应和反应物受热均匀的特性。即微波能够深入物质的内部,不依靠物质本身的热传导,因此只需要常规方法十分之一到百分之一的时间就可完成整个加热过程。
2.3 产物表征分析
2.3.1 红外分析

图2 1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的红外谱图
2.3.2 核磁共振分析
选取氘代氯仿为溶剂,对微波法合成的1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓进行核磁共振(1H-NMR)氢谱表征分析,核磁氢谱图如图3所示。

图3 1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的1HNMR图
由图可知,δ=0.56处有1个面积为4.11的双二重峰,可判断为与Si原子相连的CH2CH2Si基团的特征峰;δ=1.95附近有两个面积分别为2.19和2.10的二重峰,可判断为CH2CH2CH2基团的特征峰;δ=7.1附近面积为1.78的特征峰归属于咪唑环上活性氢原子的特征峰;δ=4.30 处有1 个面积为4.00的四重峰,可判断为与咪唑N相连的N CH2CH2基团的特征峰;δ=3.77 附近有1 个面积为12.34 的多重峰,归属于O CH2CH3基团的特征峰;δ=1.15附近有1 个面积为18.14 的二重峰,可判断为烷氧基末端的 CH3基团的特征峰。核磁共振氢谱分析可知,实验所得产品与目标产物结构中的氢环境一致,证实所得产物为1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓。
3 结论
分别采用常规方法和微波辐照法,以N-(3-乙氧基硅丙基)咪唑硅烷和3-氯丙基三乙氧基硅烷为原料,合成一种重要的有序介孔有机硅前体,即1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓;通过实验研究并分析论证了3 种常规方法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓收率差异的原因;采用微波辐照方式成功合成了1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓,且最佳合成条件是:投料N-(3-乙氧基硅丙基)咪唑硅烷4mmol,功率240W,单次微波辐照时长15s,微波辐照总时长15min,投料比n(中间体)∶n(CPTES)=1∶1.2;最佳条件下,微波辐照法合成1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的反应时间由常规法的40~45h 缩短到30min 以内,且产物收率也由81%提高到87.2%,整个反应过程无需有机溶剂和大量的冷凝水。本研究为1,3-双(3-三乙氧基硅丙基)-咪唑氯鎓的合成提供了一种高效、环保且操作简便的方法,也为双硅烷化的离子型有序介孔有机硅前体的合成提供了参考。
