儿童肾上腺危象临床特征及基因分析
2020-06-08周莛朱岷
周莛 朱岷


摘要:目的 分析肾上腺危象患儿临床特征、基因突变类型及基因型与表型的关系,提高临床对肾上腺危象患儿的认识及诊治水平。方法 回顧性分析2017年7月~2019年12月我院诊治为肾上腺危象的24例患儿的临床表现、生化检查及基因检查结果。结果 24例患儿临床表现以体重不增/倒退(95.83%)、皮肤色素沉着(91.67%)、胃肠道症状(83.33%)为主,生化检查均提示代谢性酸中毒、电解质紊乱(低钠和或高钾)、肾素活性增高,22例(91.67%)促肾上腺皮质激素(ACTH)增高,其中19例21羟化酶缺陷症17-羟孕酮增高,余5例(其他类型)17-羟孕酮无增高。共发现3种基因突变,其中19例CYP21A2基因突变、4例DAX1/NR0B1基因突变、1例STAR基因突变,其中CYP21A2基因发现1个新的剪切突变位点c.202+38dupG,DAX1/NR0B1基因发现新的移码突变c.270_280del(p.Y91Efs*18);21羟化酶缺陷症基因型与表型的相符率为84.21%。结论 儿童肾上腺危象病因复杂,临床特征缺乏特异性,遗传学有助于明确病因及预后评估判断,主张均需尽早完善基因检查。
关键词:肾上腺危象;原发性肾上腺皮质功能减退症;基因突变
中图分类号:R725 文献标识码:A DOI:10.3969/j.issn.1006-1959.2020.09.020
文章编号:1006-1959(2020)09-0067-06
Clinical Characteristics and Genetic Analysis of Children's Adrenal Crisis
ZHOU Ting,ZHU Min
(Department of Endocrinology,Children's Hospital Affiliated to Chongqing Medical University,Chongqing 400000,China)
Abstract:Objective To analyze the clinical characteristics, types of gene mutations and the relationship between genotypes and phenotypes in children with adrenal crisis, to improve the clinical understanding and diagnosis and treatment of children with adrenal crisis.Methods The clinical manifestations, biochemical and genetic examination results of 24 children diagnosed and treated as adrenal crisis in our hospital from July 2017 to December 2019 were retrospectively analyzed.Results The clinical manifestations of 24 children were mainly weight gain/regression (95.83%), skin pigmentation (91.67%), and gastrointestinal symptoms (83.33%). Biochemical examinations showed metabolic acidosis and electrolyte disturbances (low sodium and or high potassium), increased renin activity, 22 cases (91.67%) increased adrenocorticotropic hormone (ACTH), including 19 cases of 21 hydroxylase deficiency 17-hydroxyprogesterone increased,the remaining 5 cases (other types) had no increase in 17-hydroxyprogesterone. A total of 3 gene mutations were found, including 19 cases of CYP21A2 gene mutation, 4 cases of DAX1/NR0B1 gene mutation, and 1 case of STAR gene mutation, of which 1 new splicing mutation site c.202+38dupG, DAX1/NR0B1 was found in CYP21A2 gene.The gene found a new frameshift mutation c.270_280del (p.Y91Efs*18); the coincidence rate of 21 hydroxylase deficiency genotype and phenotype was 84.21%.Conclusion The etiology of adrenal crisis in children is complicated, and the clinical features lack specificity. Genetics is helpful to clarify the cause and prognosis assessment and judgment. It is suggested that genetic testing should be completed as soon as possible.
Key words:Adrenal crisis;Primary adrenocortical insufficiency;Gene mutation
兒童原发性肾上腺皮质功能减退症(primary adrenocortical insufficiency,PAI)是由于肾上腺皮质自身病变导致肾上腺皮质激素的合成和释放障碍,包括感染、出血、自身免疫性疾病、药物、遗传等因素。儿童PAI病因复杂,以遗传因素多见,其中以先天性肾上腺皮质增生症(congenital adrenocortical hyperplasia,CAH):21羟化酶缺陷症为主,比较少见的病因如3β羟类固醇脱氢酶缺乏症、DAX1基因突变、SF-1基因突变、STAR基因突变、X-连锁肾上腺脑白质发育不良等,不同基因突变类型其临床表现严重程度不一。肾上腺危象(adrenal crisis,AC)为其严重并发症,有致命性危险,临床医生需要提高警惕,早期识别及处理,但儿童期发病特别是婴儿期起病患者因其临床表现缺乏特异性,临床容易忽视或误诊为败血症、颅内感染等,导致致命性心律失常、低血糖、低血容量性休克等危及患儿生命,本研究通过回顾性分析肾上腺危象患儿临床特征及基因突变类型,提高对肾上腺危象患儿的认识及诊治水平。
1资料与方法
1.1一般资料 选取2017年7月~2019年12月在重庆医科大学附属儿童医院临床诊断为肾上腺危象的患儿24例。均符合相关指南[1,2]提出的儿童肾上腺危象定义:急性加重,不能归因于其他疾病,至少伴有以下3条中任一异常:①血流动力学改变:低血压或毛细血管充盈时间>3 s或心率增快;②至少一个显著电解质异常:高钾血症/低钠血症;③低血糖。与此同时患儿多伴有胃肠道症状(恶心、呕吐、腹痛等)、喂养困难、发热、神经系统异常(意识改变、抽搐等)。本研究获得医院伦理委员会批准,征得其患儿监护人知情同意并签署知情同意书。
1.2方法
1.2.1一般资料收集 收集患儿籍贯、性别、年龄、临床表现、血钾、血钠、血糖、pH、HCO3-、BE、ACTH、皮质醇、17羟孕酮、卧位肾素、卧位醛固酮、基因结果,部分患儿染色体核型、肾上腺CT/彩超、ACTH兴奋实验、LHRH兴奋实验、肌酶谱、血串联质谱、尿有机酸、血脂等检查结果。根据临床表现及实验室检查分为21羟化酶缺陷症、其他类型2组。
1.2.2基因测序 ①全基因组新一代测序技术(NGS)采集患儿全血(EDTA抗凝血),使用QIAamp DNA Mini试剂盒(Qiagen,中国上海,货号180134)按照生产商的说明从全血中提取基因组DNA,利用捕获芯片(MyGenostics, GenCap)将原发性肾上腺皮质功能减退症候选基因的DNA片段进行富集后再借助Illumina Next500进行高通量测序,利用BWA软件将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg19)上,利用GATK软件分析确定突变类型。②long-PCR 利用Primer primer5.0设计CYP21A2引物,对基因组DNA进行PCR扩增,扩增产物分解成长度为250~300 bp的片段并纯化,对纯化的DNA使用T4连接酶和T4碱性磷酸酶及Klenow片段进行末端修复,并在片段3端加A碱基,再通过接头连接形成标准的Solexa测序文库,利用Illumina Next 500进行高通量测序,与人类参考序列(hg19)进行比对,数据用GATK软件分析。③多重链接探针依赖扩增技术(MLPA)的检测:MLPA检测试剂盒使用迈基诺公司的CYP21A2基因文库制备试剂盒(货号:MG0017)。根据试剂盒说明书操作,数据用Gene Marker v1.6软件分析。④验证:对获取的变异位点或缺失重复利用Sanger测序或MLPA验证,并对其父母进行家系共分离验证。
1.2.3基于基因型预测临床表型模型 21羟化酶缺陷症根据临床表型严重程度分为经典型与非经典型,经典型进一步分为失盐型(salt wasting phenotype,SW)、单纯男性化型(simple virilizing phenotype,SV)。根据文献报道及相关突变体外功能学实验得到国内外广泛认同:21羟化酶缺陷症临床表型主要与不同基因突变导致酶活性降低程度不同所导致,若两个等位基因复合杂合突变,对酶活性影响最小的突变决定患儿的临床表型。将基因型分为NULL组、A组、B组、C组和D组,将大片段缺失的纯合突变、导致21羟化酶活性完全缺失如E6cluster、p.L308Ffs*6、pR357W等的纯合突变以及大片段缺失与导致酶活性完全缺失组成的复合杂合突变归为NULL组,c.293-13C>G(I2G)的纯合突变或与NULL组的突变组成的复合杂合突变归为A组;c.518T>A(p.I173N)的纯合突变或者与A组或NULL组的突变组合成复合杂合突变归为B组;对21羟化酶活性影响较小(剩余酶活性可达20%~60%)如p.V282L和p.P31L等突变,其纯合突变或者和Null组、A组、B组组成的复合杂合突变归类为C组;未确定功能的突变类型或与NULL、A组、B组、C组的突变组成的复合杂合为D组。对于NULL组、A组的基因型,预测与SW有关,容易出现肾上腺危象;B组基因型预测为SV;C组与非典型相关;D组不能预测临床表型。
1.3统计学处理 利用SPSS 23.0统计软件进行统计分析,服从正态分布的计量资料用(x±s)表示,计数资料用(n,%)表示。
2结果
2.1临床特征 患者发病年龄6 d~12.5岁,生物学性别男女比为3.8:1,分别来自四川8例、贵州8例、重庆5例,湖北3例(利川2例,恩施1例)。24例患儿临床表现为23例(95.83%)体重增长不足或倒退;22例(91.67%)有不同程度的皮肤色素沉着;20例(83.33%)有反复吐奶/呕吐、腹泻等胃肠道表现,20例中有4例伴有严重腹胀、血便,临床诊断坏死性小肠结肠炎,予以剖腹探查1例;10例(41.67%)存在性腺发育异常(女性男性化5例、男性性腺发育不良5例);11例(45.83%)低血压;1例(4.17%)意识障碍、呼吸心跳骤停;4例(16.67%)有明确家族史。
2.2辅助检查 患者实验室检查、影像学检查及染色体检查结果见表1。
2.2.1生化检查 24例(100.00%)患儿均存在明显电解质紊乱(低钠和或高钾血症),血钾(7.70±1.29) mmol/L升高明显、最高10.4 mmol/L(正常范围3.5~5.5 mmol/L),伴严重低钠血症(114.43±8.79) mmol/L,最低100 mmol/L(正常范围135~145 mmol/L),同时伴有不同程度代谢性酸中毒。其中3例(12.50%)低血糖,血糖最低0.9 mmol/L,血糖平均(5.06±2.25) mmol/L。
2.2.2内分泌实验室检查 24例中有22例(91.67%)ACTH不同程度增高;皮质醇水平减低4例(16.67%),余20例正常范围或增高;24例(100.00%)肾素活性均明显增高;醛固酮水平正常范围6例(25.00%),18例(75.00%)增高。19例21羟化酶缺陷症患儿17羟孕酮明显增高,高于检测上限;5例其他类型17羟孕酮无增高,ACTH兴奋实验无反应。病例编号22提示性激素(LH、FSH、总睾酮、雄烯二酮、脱氢表雄酮)水平低下,LHRH提示低促性腺激素性性腺功能减退症;病例编号24血脂增高,肌酸激酶增高,甘油增高。
2.2.3影像学检查 24例患儿中15例完善肾上腺影像学(肾上腺彩超/肾上腺CT),其中5例肾上腺单侧/双侧增生,3例提示肾上腺发育不良可能,余7例未见异常。
2.2.4染色体检查 10例性腺发育异常完善染色体检查,其中7例社会性别与生物学性别一致,3例社会性别与生物学性别不一致。
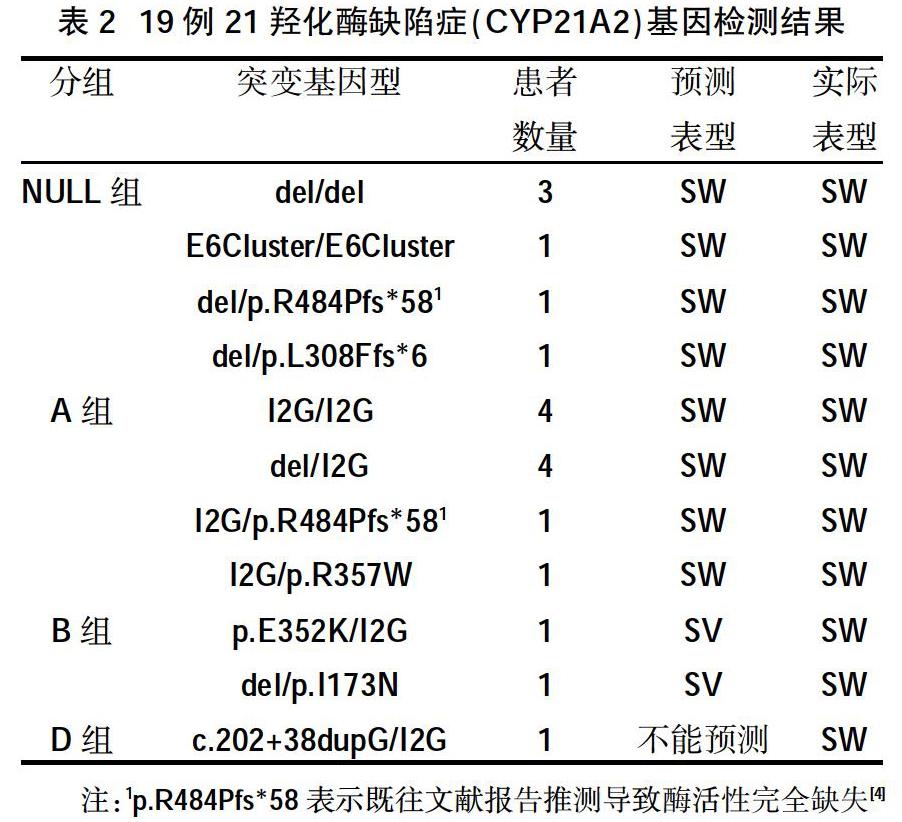
2.3基因检测结果 24例患儿完善基因检测,19例为CYP21A2基因突变,4例为DAX1/NR0B1基因突变,1例为STAR基因突变。19例CYP21A2基因突变共检测出38个等位基因,突变位点c.293-13C>G(I2G)及大片段基因缺失/转换最为常见[3,4],分别占42.11%(16/38)、34.21%(13/38),c.1451-1452delinsC(p.R484Pfs*58)、E6Cluster(p.I237N;p.V238E;p.M240K)各占5.26%(2/38),其余c.923dupT (p.L308Ffs*6)、c.202+38dupG、c.1069C>T(p.R357W)、C.1504G>A(p.E352K)、c.518T>A (p.I173N)各占2.63%(1/38)。其中c.202+38dupG为新发变异位点。STAR基因突变为c.465+2T> A(IVS4+2T>A)纯合突变,父母为携带者;DAX1/NR0B1基因突变中2例为移码突变,2例为基因缺失,见表2、表3。
2.4基因型与临床表型相符性分析 本研究中21-羟化酶缺陷症基因型与临床表型相符率为84.21%(16/19),10.53%(2/19)基因型与表型不相符,其中1例无法预测临床表型。
3讨论
肾上腺危象系肾上腺皮质功能减退症患者在感染、外伤、手术等情况下肾上腺激素分泌不足引起的一组临床症候群,临床表现缺乏特异性,容易误诊及漏诊,为PAI的严重并发症,儿童多见。本文研究中显示儿童肾上腺危象临床表现主要以体重增长不足或倒退、胃肠道症状、皮肤色素沉着为主,严重者可能出现坏死性小肠结肠炎、呼吸心跳骤停等,生化检查均提示严重电解质紊乱、代谢性酸中毒,故临床上对婴儿期存在体重不增/倒退、胃肠道症状明显,无法解释的顽固性高钾低钠血症、代谢性酸中毒,或者无明显诱因的坏死性小肠结肠炎、呼吸心跳骤停等,临床上需注意是否存在皮肤色素沉着、外生殖器畸形,同时尽早完善ACTH、皮质醇、17羟孕酮、卧位肾素+醛固酮等检查评估有无PAI可能。本文发现24例儿童肾上腺危象患儿,22例(91.67%)有ACTH不同程度增高,ACTH增高为PAI特异性表现。24例(100.00%)肾素活性增高,但醛固酮水平低下不明显,考虑与小婴儿存在生理性醛固酮抵抗及低血容量有关,故肾素活性及醛固酮水平在婴儿期及低血容量时诊断特异性不高;同时20例皮质醇水平正常范围或增高,考虑与实验室检测手段(皮质醇前体物质与皮质醇结构类似,无法区分)、残存肾上腺皮
质功能等有关,故临床上需綜合分析和鉴别。
儿童肾上腺危象以失盐型21羟化酶缺陷症为主,其次为先天性肾上腺发育不良:DAX1/NR0B1基因突变多见,与本文研究一致[5]。失盐型21羟化酶缺陷症为CAH主要类型,需与其他3β羟类固醇脱氢酶缺乏症、STAR基因突变(先天性类脂性肾上腺皮质增生症)等CAH相鉴别。19例21羟化酶缺陷症均伴有17羟孕酮异常增高,而LCAH17羟孕酮无增高,ACTH兴奋试验无反应,为其鉴别要点。经典型3β羟类固醇脱氢酶缺乏症部分女性患儿因外周Ⅰ型3β羟类固醇脱氢酶可将部分17羟孕烯醇酮和脱氢表雄酮转化为17羟孕酮和少量雄激素,导致17羟孕酮升高至21-羟化酶缺陷症水平,临床难以区分;AHC以DAX1/NR0B1基因突变多见,与LCAH、SF-1基因突变、P450scc缺陷等均导致所有肾上腺类固醇激素合成障碍,17羟孕酮无增高,ACTH兴奋实验无反应,诊断困难,均需要进一步基因分析明确诊断。
本研究19例失盐型21-羟化酶缺陷症患儿主要来自西南地区,最常见类型为点突变c.293-13C>G(I2G)42.11%,其次为大片段基因缺失/转换34.21%,与文献报道基本一致[4,6,7],其中中国北京地区失盐型患儿以大片段基因缺失/转换出现的频率最多[8],中国不同地区可能存在差异。本研究中,21-羟化酶缺陷症基因型与表型相符率为84.21%,提示基因型与表型相符性较好,其中本文患儿c.202+38dupG/I2G复合杂合突变,临床表现为失盐型,c.202+38dupG为新发突变位点,系剪接突变,位于内含子上,可引起mRNA的剪切异常,导致相应蛋白质的功能改变且不属于多态性位点,在人群中发生频率极低,根据突变分析推测c.202+38dupG系致病位点且导致21羟化酶活性完全缺失。通过基因型与表型相关性分析,临床上可通过临床表型评估验证基因型,基因型也可预测临床表型的严重程度,为后续临床诊疗及随访提供重要参考依据。然而仍有10.53%的基因型与表型不相符,基因型预测为单纯男性化型,但基因型del/I173N也有文献报道为失盐型[4],具体机制不详,是否不同大片段缺失重复与I173N复合杂合导致21羟化酶活性不同,也为进一步的研究提供方向。
DAX1/NR0B1基因突变是引起儿童PAI较常见的病因,位于Xp21.3,X连锁隐性遗传病,因有不同的遗传外显性,发病年龄早晚、临床表现均有明显差异,基因型与表型相关性尚不明确,同时Xp21区域含有多个疾病位点如 IL1RAPL1、NR0B1、GK和DMD。若DAX1/NR0B1基因大片段缺失,除累及肾上腺功能外,还可能合并甘油激酶缺乏症、杜氏肌营养不良等,临床上统称为Xp21邻近基因缺失综合征。本研究中有1例为8-79号外显子>3.9Mb缺失,虽无相应临床表现,实验室检查提示甘油激酶缺乏症、血脂及肌酸激酶增高,故临床上疑诊AHC男性患儿,特别针对基因突变提示大片段缺失患儿还应关注神经系统发育、肌酶谱、血脂、血尿质谱分析等[9];同时此类患儿大多数存在不同程度性腺发育落后,本文中有1例患儿12.5岁就诊时发现睾丸容积小,骨龄CHN评分831分,性激素水平低下,临床对于此类型患儿需密切观察性腺发育情况,根据病情严重程度酌情给予雄激素替代治疗促进男性化发育。
较少见的儿童PAI病因:STAR基因突变,系常染色体隐性遗传,位于8p11.2,两性均表现为女性外阴,临床难以鉴别,需依靠分子检测手段。同时有文献显示若变异蛋白活性低于野生型的10%,临床症状多为经典型LCAH;若活性保持在10%~25%,则临床表型可呈非典型LCAH[10],本研究STAR基因突变为c.465+2T> A(IVS4+2T>A)纯合突变,既往文献推测该剪切突变为致病性突变[11,12],可导致严重临床表型,基因推测为经典型。目前样本量少,基因型及表型的相关性需更大样本及功能试验分析研究。
综上所述,儿童肾上腺危象以失盐型21-羟化酶缺陷症多见,其次为AHC(DAX1/NR0B1基因突变),临床表现以皮肤色素沉着、胃肠道症状、体重不增/倒退为主,21羟化酶缺陷症以17羟孕酮增高为主,AHC及LCAH 17羟孕酮无增高,为临床主要鉴别要点,但因疾病临床表现缺乏特异性,存在实验误差,不同类型PAI的预后及随访内容所有差异,故主张尽早完善基因检查,明确病因,判断预后,制定不同的随访方案。21羟化酶缺陷症有良好的基因型与临床表型关系,可通过临床表型评估验证基因型,基因型也可预测临床表型的严重程度,为后续临床诊疗及随访提供重要参考依据;LCAH基因型及表型的关系需更大样本的分析研究。
参考文献:
[1]Bornstein SR,Bruno A,Wiebke A,et al.Diagnosis and Treatment of Primary Adrenal Insufficiency:An Endocrine Society Clinical Practice Guideline[J].The Journal of Clinical Endocrinology&Metabolism,2016,101(2):364-389.
[2]Louise RR,David JT,Constantine AS,et al.Adrenal Crises in Children:Perspectives and Research Directions[J].Hormone Research in Paediatrics,2017,55(2):1-10.
[3]Wang R,Yu Y,Ye J,et al.21-hydroxylase deficiency-induced congenital adrenal hyperplasia in 230 Chinese patients:Genotype-phenotype correlation and identification of nine novel mutations[J].Steroids,2016(108):47-55.
[4]Ling S,Xi Y,Jing C,et al.Clinical presentation and mutational spectrum in a series of 166 patients with classical 21-hydroxylase deficiency from South China[J].Clinica Chimica Acta,2018(486):142-150.
[5]Eyal O,Levin Y,Oren A,et al.Adrenal crises in children with adrenal insufficiency:epidemiology and risk factors[J].European Journal of Pediatrics,2019.
[6]舒剑波,张新杰,徐晓薇,等.21-羟化酶缺乏症CYP21A2基因突变及表型分析[J].中华内分泌代谢杂志,2019,35(1):21-25.
[7]Mendes C,Matos IV,Luís Ribeiro,et al.Congenital Adrenal Hyperplasia Due to 21-Hydroxylase Deficiency:Genotype-Phenotype Correlation[J].Acta Medica Portuguesa,2015,28(1):56-62.
[8]Bo Z,Lin L,Zhao LL.Molecular diagnosis of Chinese patients with 21-hydroxylase deficiency and analysis of genotype–phenotype correlations[J].Journal of International Medical Research,2017,45(2):481-492.
[9]Heide S,Afenjar A,Edery P,et al.Xp21 deletion in female patients with intellectual disability:Two new cases and a review of the literature[J].European Journal of Medical Genetics,2015,58(6-7):341-345.
[10]Miller WL,Bose HS.Early steps in steroidogenesis:Intracellular cholesterol trafficking[J].Journal of Lipid Research,2011,52(12):2111-2135.
[11]Camats N,Pandey AV,Fernández-Cancio M,et al.STAR splicing mutations cause the severe phenotype of lipoid congenital adrenal hyperplasia:insights from a novel splice mutation and review of reported cases[J].Clinical Endocrinology,2014,80(2):191-199.
[12]謝婷,郑纪鹏,黄永兰,等.先天性类脂质性肾上腺皮质增生症临床特点及StAR基因突变分析[J].中国当代儿科杂志,2015(5):472-476.
收稿日期:2020-03-17;修回日期:2020-04-08
编辑/成森
作者简介:周莛(1987.1-),女,重庆人,硕士,住院医师,主要从事内分泌及代谢性疾病研究
通讯作者:朱岷(1963.3-),女,重庆人,硕士,主任医师,教授,硕士生导师,主要从事小儿生长发育、肾上腺疾病及性发育异常研究
