宏基因组测序技术在传统酿造食品微生物群落分析中应用研究进展
2020-06-08王文平续丹丹
高 航,张 欣,赵 燕*,王文平,续丹丹,王 鹏
(1.北京食品科学研究院,北京 100068;2.北京市食品酿造所,北京 100050)
我国传统酿造食品历史悠久,种类丰富,以其独特的魅力广受欢迎。酱油、酱类、食醋、腐乳、白酒和泡菜等都是最常见的传统酿造食品,它们是以谷类、豆类或蔬菜等为原料,将自然界的微生物引入发酵过程中共同作用而形成风味独特、营养价值较高的食品[1]。
传统酿造技术多采用固态或半固态发酵工艺,在开放或半开放式的环境下,通过微生物群落的盛衰交替和协同作用酿制食品的过程[2-4]。经过漫长的驯化与筛选,不同地区的酿造食品形成了较为稳定的工艺与微生物群落,赋予了酿造食品独特的风味、滋味和丰富的营养。研究表明,醋酸菌产生以醋酸为主的有机酸,对高脂饮食诱导的肥胖小鼠具有良好的抗肥胖和抗炎活性[5];黄酒酿造过程中以霉菌和酵母菌为主代谢产生的功能性多糖,可以抑制小鼠的淋巴细胞增殖而增强免疫抑制小鼠的免疫活性[6];在研究传统酿造酱油的生理活性时发现,以1.8 mL/kg的剂量灌胃高尿酸大鼠30 d,其血清尿酸水平及黄嘌呤氧化酶得到了良好的抑制,缓解了高尿酸症状[7]。在系统地剖析酿造过程中微生物群落的基础上,深入挖掘功能基因和微生物间的相互作用关系,能更充分地改善与提升酿造食品的品质和营养[8-9]。近年来,应用不同生物技术研究酿造食品中的微生物群落成为了热点[10-11],其结果为开发出“色、香、味、营养”俱全的酿造食品奠定理论基础。
在微生物学研究领域中,99%以上的微生物都是目前未(难)被纯培养的,这对于具有高度物种多样性的传统酿造食品而言,进一步了解其微生物群落结构和功能具有更大的挑战[12]。随着分子生物学技术的快速发展,传统酿造食品微生物群落结构的研究已不仅仅局限于传统的可分离培养微生物研究[13]。高通量测序技术的更新、测序成本的降低以及测序通量的扩展等优势不断加强,为直接分析群落中全部微生物的种群组成、分布及其动态演替提供了有利的技术支撑[14-15]。宏基因组测序技术不仅提供了更为丰富的微生物物种的基因信息,而且还有效提高了检测效率和结果准确性,为研究者更全面地解析微生物群落结构、变化规律并挖掘潜在的功能基因提供契机,从而得以进一步开展对传统酿造食品的深入探索[16]。本文将对宏基因组测序技术在传统酿造食品微生物群落研究中的应用进展进行综述,并分析讨论其面临的主要问题和发展趋势,为酿造食品的科学研究和工业生产提供相关理论基础。
1 宏基因组测序技术
1.1 宏基因组测序技术概述
1991年,DELONG E F等[17]首次提出环境基因组学(environmental genomics)的概念,在对太平洋浮游生物进行系统分类时,通过直接提取脱氧核糖核酸(deoxyribonucleic acid,DNA)并构建基因文库,发现了15种全新的细菌序列,这是第一次不依赖于纯培养的基因组分析而发现新物种的报道。1998年,HANDELSMAN J等[18]研究土壤微生物群落时首次将“特定环境中全部微小生物遗传物质的总和”定义为宏基因组(metagenome)。目前,宏基因组的定义又被更正为特定环境中细菌和真菌遗传物质的总和。宏基因组测序技术正是以高通量测序为依托,通过样品和基因(组)的富集、提取特定环境中的基因组DNA、构建宏基因组文库和筛选目的基因等流程来研究微生物的方法,从而达到研究宏基因组的目的[19](见图1)。
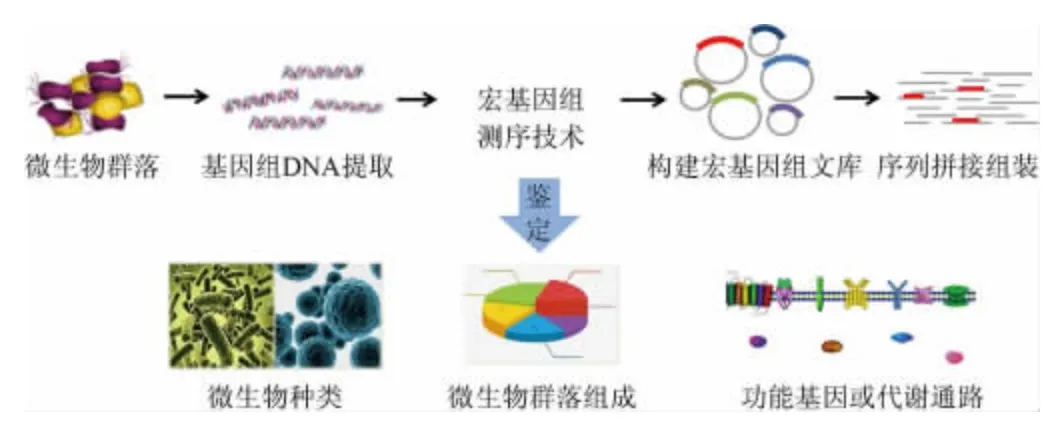
图1 宏基因组测序技术流程Fig.1 Sequencing process of metagenomics
宏基因组测序技术的结果除了包含群落中各种微生物的分类信息以外,更包含了所有微生物的基因信息。随着高通量测序成本的逐渐降低,宏基因组测序技术也因而应用在更多领域的微生物研究中,包括对大量不可培养的微生物,以及新的微生物或功能基因的认知,极大地增加了人类对不可培养微生物群落的组成、演替、功能及其互相作用的认识,开辟了微生物与环境或微生物与微生物之间的相互作用关系研究的新方法。
1.2 高通量测序技术在宏基因组测序中的应用
高通量测序技术是现今应用最广泛的测序技术,其特点是成本低、通量高、速度快,可以快速产生大量的数据[20]。1977年Sanger测序技术诞生并在“人类基因组计划”中被广泛使用,被称为“第一代测序技术”[21]。第一代测序技术具有读长较长、准确率较高的特点,但其通量较低,分析时间较长等无法满足大批量的数据分析。第二代测序技术依托的高通量测序平台,包括Illumina公司的Solexa Genoma Analyzer测序平台、罗氏公司的454GS FIX测序平台和ABI公司的SOLiD测序平台,它们的测序深度可以在一定程度上弥补读长较短所带来的问题,深入并且快速的测序过程也使它们得以成为现今应用最广泛的测序技术[22]。而第三代测序技术是一种集高通量、快速度、读长更长及低成本等多种优点于一身的新型测序技术,它的出现为宏基因组学的研究注入了新活力[23]。
1.2.1 基于宏基因组测序技术的微生物群落分析
在原核微生物中,与23SrRNA和5SrRNA相比,16SrRNA包含适量且足够的信息量和序列长度,因此作为研究物种进化关系的“黄金标准”[24]。16S rRNA的微生物群落分析可产生深度的16S rRNA测序数据,并通过比对或聚类的分析方法,将16S rRNA序列聚类成分类操作单元(operational taxonomic unit,OTU),利用OTU的数目、各个OTU的序列数来分析估计物种多样性和丰度,从而实现对数据来源的微生物物种和微生物群落的物种构成进行分析[25]。在真核微生物中,真菌DNA的碱基组成遗传稳定,不易受环境影响,而且在其生长的任何阶段均可获得。内部转录间隔区(internal transcribed spacer,ITS)在不同菌株间存在丰富的变异,对ITS区进行序列分析丰富了真核生物核糖体基因ITS的序列信息。ITS1和ITS2是中度保守区域,其保守性基本上表现为种内相对一致,种间差异比较明显,通过对ITS序列进行DNA测序,进而将测序得到的ITS序列与已知真菌ITS序列比较,为真菌分类鉴定等研究提供了十分重要的资料和依据[26]。但是,在鉴定物种方面,16S rRNA和ITS的分类精度是有限的,一般仅能够精确分类到属水平,其序列很多注释不到种水平。其原因是16S rRNA和ITS测序技术能够区分非常相近的高变区的特异性序列片段有可能不在扩增区域内,即非全长的可变区序列覆盖范围不够导致无法鉴定到种[27]。然而,同一种的不同个体在基因组信息上也会有差异,只分类到属是远远不够的。
宏基因组测序技术是先将微生物基因组DNA随机打断成若干条500 bp的小片段,并在片段两端加上通用引物进行PCR扩增测序,然后再进行组装拼接得到基因序列,众多基因即可构成完整的基因组[28]。因此,在物种鉴定过程中,宏基因组测序技术并不受高突变区的影响,具有明显的优势。此外,宏基因组测序得到的微生物群落基因信息,还可进一步开展特定环境中微生物群体基因组成及功能分析、微生物群体的多样性与丰度演替变化等研究,为深入分析微生物与环境、微生物与宿主之间的关系以及挖掘具有特定功能的基因提供有力的支撑[29]。
宏基因组测序无需分离纯培养微生物,直接对样品中微生物群落的总基因进行测序和分析,可同时鉴定出细菌和真菌,不仅保证了低丰度的微生物可以被检测出来,而且检测效率高,结果准确,较大地扩展了微生物资源的利用,为复杂的微生物群落体系研究提供了有效工具[30]。宏基因组深度测序可以揭示或估计环境中真实的物种多样性和遗传多样性,挖掘具有应用价值的基因资源,应用于开发新的微生物活性物质。目前,宏基因组测序技术已经应用在食品微生物[31]、肠道微生物[32]等多领域的微生物群落研究中。
1.2.2 基于宏基因组测序技术的功能基因分析
基于16S rRNA和ITS测序技术只能依赖于基因库中已知基因序列进行分析,而缺乏具体的功能基因信息。相比之下,宏基因组测序技术在一定程度上弥补了这些缺陷,使得人们可以更系统地研究微生物群落结构及其功能基因,全面分析其组成、变化规律、亲缘进化并挖掘出潜力丰富的功能基因,从而得以进一步揭开酿造食品微生物群落的奥秘[33]。
功能基因的筛选无需依赖于已知的基因信息,因此可能得到新的基因和产物;但其受制于外源基因在宿主细胞中的表达,并因检测手段的局限性和有效性只能在大量分析的基础上筛选出极少的具有活性的基因。鉴于此,发展高通量、高灵敏度、快速简便的筛选方法或体系十分必要[34]。高通量测序数据首先映射到微生物基因组和基因的已知数据库(如美国国家生物技术信息中心(National Center of Biotechnology Information,NCBI)数据库或京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)数据库),并获得每个宏基因组样本的基因目录,显然此种方法需要有足够的数据库支撑[35]。针对宏基因组测序技术的数据分析,一般分为基于比对的方法和不基于比对的方法,二者均通过对基因丰度水平的估计实现对基因进行预测或者功能性分析。基于比对的序列比较方法通常先被映射到已知的基因或者生化路径代谢数据库中,再对基因或基因家族的丰度水平进行估计,最后结合一些功能基因、代谢通路、信号通路等数据库,对研究者感兴趣的部分进行分析或进行基因预测[36]。不基于比对方法克服了基于比对的方法对有限的参考数据库的依赖,其通过统计长度为k的元组出现的频度而计算,在未知物种为主的宏基因组分析中,以长度k分析的优势非常明显,是宏基因组的一个重要的研究方向[37]。
2 宏基因组测序技术在传统酿造食品中的应用
2.1 传统酿造食品中微生物群落结构的解析
2.1.1 微生物群落组成分析
传统的酿造食品多在开放式环境中经混菌发酵形成,其中富含复杂多样的微生物群落结构体系[38]。目前,只有少部分优势菌种通过纯培养技术鉴定出来,而基于16S rRNA克隆建库[39]、限制性片段长度多态性(restriction fragment length polymorphism,RFLP)[40]、聚合酶链式反应-变性梯度凝胶电泳(polymerase chain reaction-denaturing gradient gel electrophoresis,PCR-DGGE)[41]等无需纯培养的分析技术,也只能鉴定出丰度较高或参考数据库中可供参考的基因信息。更全面地了解不同发酵阶段的微生物群落组成,是提高酿造食品品质和发酵效率的必要条件。因此,基于宏基因组测序技术能鉴定出更低丰度的微生物群落及精准鉴定到种级别的优势上,广泛地应用在更全面和更微小的微生物群落体系研究中。
在印度米酒酒曲(Xaj-pitha)的研究中,BORA S S等[42]首次应用宏基因组测序技术揭示了更为全面的微生物群落特征,其中鉴定出的微生物包括以乳酸菌为主的细菌、根霉、毛霉和曲霉等产淀粉酶菌株;以季也蒙毕赤酵母(Meyerozyma guilliermondii),威克汉姆西弗酵母(Wickerhamomyces ciferrii)和酿酒酵母(Saccharomyces cerevisiae)为主的产乙醇菌株等。但对于所获得的宏基因组数据,作者仅报道了分类学结果,并没有进一步分析功能基因组和代谢途径。NIE Z等[43]应用宏基因组测序技术在传统食醋中首次发现了相对丰度不到0.1%的念珠藻属(Nostoc),类诺卡氏菌属(Nocardioides),丙酸菌属(Propionibacterium),克雷伯氏菌属(Klebsiella)和片球菌属(Pediococcus)。在工业生产中,念珠藻属可产生200多种次生代谢物,被用于蛋白质、维生素和多不饱和脂肪酸的生物来源[44];而丙酸菌属的发酵也可大量生产维生素B12[45]。此类菌种虽然丰度较低,但是功能却不可忽视,尤其通过其在工业生产中的作用可推测在食醋的生产发酵中可能具有类似作用。对比前人通过培养方式,仅能分离鉴定出食醋在发酵过程中的醋酸菌(主要包括醋杆菌属、葡萄糖杆菌属和葡萄糖醋酸杆菌属)、乳酸菌(主要包括乳杆菌属和乳球菌属)和芽孢杆菌属[46],而宏基因组测序技术大大提高了微生物菌种的覆盖率。类似地,孙炜宁等[47]利用宏基因组测序技术对东北酸菜进行了深入研究,应用宏基因组测序技术首次在东北酸菜中鉴定出属于肠杆菌的果胶杆菌(Pectobacterium)和拉恩氏菌(Rahnella)。在食品安全方面,肠杆菌可能导致食品污染,甚至可能引发的食源性疾病。酸菜中肠杆菌的存在主要与酸菜直接使用未清洗的白菜作为腌制材料以及低盐浓度(0.5%~1.0%)的发酵液有密切的关系。因此,在酸菜发酵中检测和控制发酵体系中肠杆菌科细菌的丰度和动态变化情况有助于积极控制和消除发酵蔬菜中亚硝酸盐和生物胺带来的危害。唐婧等[48]分别提取2011-2013年的茅台酒酒曲微生物总基因组DNA,基于宏基因组测序技术对茅台酒酒曲细菌群落多样性进行了研究。结果表明,茅台酒酒曲微生物群落构成稳定,主要分布于γ-变形菌纲(50%以上)和芽孢杆菌纲(30%以上),分属于魏斯氏菌、片球菌、明串球菌、糖多孢菌、欧文氏菌、真丝菌、短状杆菌、鞘氨醇杆菌、醋酸杆菌、糖单孢菌、盐单胞菌和德库菌。因此,宏基因组测序技术的应用在保证茅台酒的微生物群落稳定方面提供了一定的技术支撑。
2.1.2 微生物菌群动态演替分析
宏基因组测序技术不仅可以快速准确地解析微生物群落的结构,并且可以同步确定各种微生物的相对丰度变化水平[49]。酿造食品通过微生物菌群的代谢生成各类物质,赋予酿造食品不同的风味和营养功能[50]。在酿造过程中,菌群结构的动态演替直接影响代谢产物的形成,微生态调控被视为改善传统酿造食品风味和营养物质的关键与核心技术[51]。聂志强等[52]应用宏基因组测序技术对天津独流老醋醋酸发酵过程中的群落变化研究中发现,在醋酸发酵过程中,醋醅中一些相对丰度较低的细菌(相对丰度均在0.01%以上)也发生着较大的变化。其中,类诺卡氏菌属(Nocardioides)的相对丰度呈上升的趋势,在末期相对丰度达到0.06%,说明其具有较强的醋酸耐受性。假单胞菌属(Pseudomonas)随着醋酸发酵的进行相对丰度从0.14%下降到0.04%后逐渐消失,说明醋酸对假单胞菌属菌的影响较大。另外,埃希氏杆菌属在发酵前期到中期呈增加的趋势,而克雷伯氏菌属却仅出现在发酵末期,说明克雷伯氏菌有一定醋酸耐受性。在整个发酵周期中,相对丰度>0.01%的细菌达到10种,说明食醋固态酿造过程中来源于样品和环境中的多种微生物能够自发地繁殖、富集,这也导致了传统食醋丰富的风味。
在研究传统酿造酱油的微生物群落过程中,WEI C L等[53]采用PCR-DGGE法研究发现,在酱油的整个发酵过程中,以四种葡萄球菌属(Staphylococcus)和四种杆菌属(Bacillus)为优势菌,库特氏菌(Kurthia)和克雷伯氏(Klebsiella)在制曲阶段出现,在糖化阶段逐渐消失。WANG H等[54]基于宏基因组学技术,比较了酱油发酵中制曲和糖化阶段的细菌群落变化。制曲阶段共鉴定出29属细菌,后期7个属消失;糖化阶段鉴定出34属细菌,后期又出现了12个新的细菌属。制曲和糖化阶段的优势菌种分别为库特氏菌(Kurthia)、魏斯氏菌属(Weissella)、葡萄球菌(Staphylococcus)和葡萄球菌(Staphylococcus)、库特氏菌(Kurthia)、肠球菌(Enterococcus)、明串珠菌(Leuconostoc)。在制曲和糖化阶段,含量仅有0.21%的四体球菌属(Tetragenococcus)也同样被鉴定出来。通过比较,DGGE法与宏基因组测序法得到的结果在微生物丰度方面相似。显然,宏基因组测序法比其他研究方法获得了更高的物种覆盖率,从而全面阐明酿造过程中微生物群落的动态变化。
宏基因组测序技术的另一大优势即为可同时鉴定出真菌和细菌的群落变化,这一特点大大提高了检测效率,同时也减少了因方法选择的局限性带来某部分微生物物种信息的缺失。因此,宏基因组学技术的应用受到越来越多研究者的青睐。XU D D等[55]在研究腐乳发酵过程中真菌和细菌群落的动态变化时发现,腐乳发酵前期以毛孢菌属、放线菌属和隐球菌属为主,发酵后期以红曲霉属和曲霉属为主。此外,在加入混合料后首次检测到芽孢杆菌,但在发酵结束时,芽孢杆菌数量急剧下降至极低水平(0.07%)。因此,发酵过程中各个阶段微生物动态变化的研究结果可进一步实现对腐乳安全性和质量的评价。
2.2 传统酿造食品中功能微生物或功能基因的鉴定
中国传统酿造食品中微生物物种的高度多样性使得进一步分析微生物群落的代谢功能成为一个更大的挑战。功能微生物和功能基因的鉴定有助于更清晰地在众多微生物菌群中锚定对产品品质或功能起到重要作用的微生物群落或基因,以便能更准确地进行分析研究[56]。WU L等[57]采用宏基因组测序技术对香醋的微生物群落结构和功能微生物进行了研究,进而了解与食醋风味相关的微生物菌群。结果显示,食醋中细菌、真核生物和古细菌的相对丰度分别为91.75%、0.19%和8.06%。此外,共有来自951属细菌的蛋白编码基因被鉴定出来,其中42.3%与代谢通路相关,28.3%与遗传信息相关,还有10.1%与环境信息相关。食醋的风味研究结果表明,乙偶姻是在醋酸发酵过程中提高香醋风味的重要物质,其合成受到多种代谢途径的调控[58]。然而,在以往的研究中,探索产生乙偶姻的具体微生物并未被清晰的识别出来,而宏基因组测序技术的应用可在已知的乙偶姻代谢途径中瞄定功能微生物进行深入研究。LU Z M等[59]通过宏基因组测序技术,在物种水平上揭示了乙偶姻代谢途径中微生物的分布差异。乙偶姻代谢途径主要是在乙酰乳酸合酶和乙酰乳酸脱羧酶共同调控下完成。结果显示,乙酰乳酸合酶主要由布氏乳杆菌(L.buchneri)(21.0%)和巴氏杆菌(A.pasteurianus)(29.3%)产生;而布氏乳杆菌(L.buchneri)(26.5%)、罗伊氏乳杆菌(Lactobacillus reuteri)(11.6%)、巴氏杆菌(A.pasteurianus)(5.6%)、发酵乳杆菌(L.fermentum)(3.2%)和短乳杆菌(L.brevis)(2.8%)参与了乙酰乳酸脱羧酶的合成代谢。双乙酰是合成乙偶姻主要的前体物质,巴氏醋杆菌(Acetobacter pasteurianus)和四种乳酸菌,包括布氏乳杆菌(Lactobacillus buchneri)、罗伊氏乳杆菌(Lactobacillus reuteri)、发酵乳杆菌(Lactobacillus fermentum)和短乳杆菌(Lactobacillus brevis)是醋酸发酵过程中促使双乙酰向乙偶姻转化的功能微生物菌种。GUO M等[60]在研究中国传统酿造白酒时发现,真核生物占整个微生物群落的比例很小[(6.0±1.4)%]。但它们对白酒发酵过程中代谢产生乙醇有重要作用,半乳酵母菌能产生高活性的纤维素酶,以有效地进行酶的糖化。乳杆菌(Lactobacillus)、互养单胞菌(Syntrophomonas)、甲烷囊菌(Methanoculleus)、甲烷杆菌属(Methanobacterium)、芽孢杆菌(Bacillus)、梭状杆菌(Clostridium)、耐碱酵母属(Galactomyces)、念珠菌(Candida)、毕赤酵母(Pichia)、青霉菌(Penicillium)和曲霉菌(Aspergillus)是白酒中的活性微生物群落,这些微生物也与白酒中主要的风味成分形成(己酸乙酯、乳酸乙酯、乙酸乙酯等)所一致。此类微生物与乳酸、乙醇、丁酸、乳酸乙酯、短链脂肪酸、乙酯的生成有直接关系。从某种意义上说,白酒的风味化合物微生物群落结构密切相关。PARK J M等[61]研究泡菜29 d发酵过程的宏基因组,揭示了一系列碳水化合物异养乳酸发酵的相关基因,这与检测到发酵产物甘露醇、乳酸、乙酸乙酯、乙醇等相吻合。将宏基因组序列与不同菌属微生物基因组序列比较发现,大部分的宏基因组测序序列与肠膜明串珠菌(Leuconostoc mesenteroides)和沙克乳酸杆菌(Lactobacillus sakei)的基因序列具有高度相似性,表明泡菜发酵过程中这两种菌很可能发挥了重要作用。
2.3 传统酿造食品中微生物与环境的相互作用关系
中国传统发酵食品开放程度高、微生物构成复杂,发酵过程并非单个微生物的叠加,还存在复杂的交互作用机制,包括细胞与细胞之间的物理接触、代谢物交换、水平基因转移等方式,这些多种多样的作用方式最终形成了微生物之间的共生、竞争、偏利共生及偏害共生等交互作用关系,进而影响了群落整体的组成结构以及功能[62]。除微生物群落内部的互作以外,微生物与环境之间也有复杂的交互作用关系。随着发酵过程的推进,酿造微生态环境也不断改变,进而又作用于微生物群落,影响着群落的结构和功能,并最终影响产品品质[63]。因此,系统理解传统酿造食品的发酵进程,需要明晰微生物与环境之间的相互作用关系,才能有效建立调控群落功能的策略。
在中国传统的白酒酿造过程中,来自于原材料、大曲及窖泥等环境中的各种微生物已逐渐形成了稳定的微生物群落结构,从而对白酒品质和风味产生重要的影响。GUO M等[60]应用宏基因组测序技术研究了来自泸州、绵阳和成都三地的白酒品质与环境的相互作用规律。结果显示,在酿造过程中,酒的品质特征与产地微生物群落高度相关。不同的地理环境促使窖泥累积了不同微生物群落,进一步影响了白酒品质和风味。泸州、绵阳和成都三地的窖泥中含有共同菌种133属,而泸州地区的窖泥还检测出了188个特异属,绵阳和成都地区仅有29和13属特异菌,这些菌属发酵产生不同强度的己酸乙酯、乳酸乙酯、乙酸乙酯等,赋予了终产品不同的特征风味。在对三种白酒的挥发性成分分析时表明,不同产地的白酒表现出明显的感官特征区别,泸州地区的丰富微生物群落促使了优质白酒的生产。因此,深入研究不同产地的产品中微生物群落差异,有助于明确地域因素对酿造食品品质的影响。
3 展望
近年来,宏基因组测序技术在分析传统酿造食品的微生物群落组成、微生物群落的动态演替、功能基因或功能微生物以及微生物与环境的相互作用关系中取得了一些进展,为酿造食品的深入研究或工业生产都起到了一定的积极作用。然而,宏基因组测序技术的应用依旧存在着一些不可避免的局限性,如宏基因组测序数据量较大,分析有一定的难度;测序分析费用较高,大大增加了样品重复性试验的成本。随着测序技术的不断发展,三代测序技术如PacBio RS很好地弥补了第一、二代测序技术的不足,为宏基因组测序技术更准确、更便捷的应用提供了更有力的技术支撑。
宏基因组测序技术在研究微生物多样性或明确功能基因方面都有无可代替的作用,但是在微生物的巨大体系中,信使核糖核酸(messenger ribonucleic acid,mRNA)、核糖体核糖核酸(ribosome ribonucleic acid,rRNA)、转运核糖核酸(transfer ribonucleic acid,tRNA)、非编码核糖核酸(noncoding ribonucleic acid,ncRNA)以及蛋白质均是行使细胞功能的重要承担者。因此,在获取微生物基因信息的基础上,进一步明确微生物的mRNA和蛋白质表达,才能准确描述微生物的功能和状态。宏基因组学与宏转录组学、代谢组学与蛋白质组学等多组学技术的联用将有助于更清晰地了解传统酿造食品中微生物的群落结构演替、相互作用、功能菌株对食品品质和风味的贡献,并挖掘出新的功能基因。未来,生物信息学、分子生物学等多学科理论知识的丰富以及更新一代测序技术、更加完善的基因数据库、先进的数据分析工具的应用以及多种组学方法的结合将给酿造食品微生物的研究带来新的曙光。
