SPE-HPLC-MS/MS法同时测定电子烟烟液中9种甜味剂
2020-06-06廖惠云马梦婕朱怀远陈晶波朱龙杰袁益来
廖惠云,吴 洋,马梦婕,朱怀远,曹 毅,陈晶波,张 华,朱龙杰,张 媛,袁益来
江苏中烟工业有限责任公司,南京市建邺区兴隆大街29 号 210019
自2004 年以来,电子烟经过发展已具备一定规模[1-2]。电子烟烟液是电子烟的重要组成部分,其主要成分是含烟碱的丙二醇、甘油或乙二醇等,同时为满足不同抽吸者的需求,亦会添加各种香精[3-5]。文献报道在电子烟烟液等新型烟草制品中检出甜味剂[6-7]。甜味剂是赋予食品甜味的功能性食品添加剂。我国食品添加剂相关标准允许使用10 余种合成甜味剂,并规定了其在不同食品中的使用限量[8]。合理使用人工合成食品甜味剂是安全的;但如果超限量、超范围,则会对消费者的健康产生一定负面影响[9]。因此,建立具有高通量、高准确度、高灵敏度的甜味剂检测方法,加强电子烟烟液添加剂使用监管就显得尤为必要。
目前,对于甜味剂的测定主要集中于饮料、白酒、乳制品等[10-11],分析方法分别有液相色谱法[12-13]、离子色谱法[14-15]、毛细管电泳法[16-17]、气相色谱法[18-20]等,以液相色谱法为主。因不同甜味剂理化性质、电化学性质差异较大,液相色谱法很难满足多种甜味剂同时检测的需求,离子色谱法、毛细管电泳法等也因灵敏度低等原因应用不多,因而有必要开发简单快速、灵敏度高的检测方法。HPLC-MS/MS 技术由于具有检测通量大、准确灵敏的特点,被广泛用于食品领域的检测,应用该技术同时分析多种甜味剂已经成为一种趋势[11]。考虑到食品工业对甜味剂的使用大多以复配形式添加,在相关研究基础上[6-7,21],本研究中建立了固相萃取-液相色谱/串联质谱(SPE-HPLC-MS/MS)法同时测定电子烟烟液中安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、阿力甜、新橙皮甙二氢查尔酮、纽甜和甜菊糖苷9 种甜味剂的质量分数,旨在为电子烟烟液中甜味剂的检测提供方法参考。
1 材料与方法
1.1 材料、试剂和仪器
8 种不同品牌市售电子烟烟液,编号为1#~8#,其中1#~6#为烟草原味,7#为薄荷味,8#为水果香味。
安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、阿力甜、新橙皮甙二氢查尔酮、纽甜和甜菊糖苷(标准品,≥98.0%,美国Aladdin 公司);华法林钠(内标,≥98.0%,浙江玛雅试剂有限公司);甲醇、乙腈(HPLC 级,美国Tedia 公司);甲酸(色谱纯)、乙酸铵(AR)(国药集团化学试剂有限公司)。
1260/6460超高效液相色谱-三重四极杆质谱仪(配备电喷雾电离源,美国Agilent公司);KQ-500DE超声波发生器(昆山市超声仪器有限公司);EVA08 全自动定量浓缩仪(北京普立泰科仪器有限公司);T201 电子天平(感量0.000 1 g,瑞士Mettler Toledo 公司);Milli-Q 超纯水系统(美国Millipore 公司);C18硅胶键合固相萃取柱和Oasis HLB 固相萃取柱(500 mg/6 mL,美国Waters 公司)。
1.2 方法
1.2.1 标准工作溶液的配制
(1)内标溶液和标准储备液
准确称量5 mg 华法林钠(精确至0.1 mg),用水溶解并转移至10 mL 容量瓶中,用超纯水定容,配制成浓度为500µg/mL 的内标溶液。分别准确称取6 mg 安赛蜜、14 mg 糖精钠、6 mg 甜蜜素、14 mg 三氯蔗糖、3 mg 阿斯巴甜、3 mg 阿力甜、12 mg新橙皮苷、1 mg 纽甜、10 mg 甜菊糖(精确至0.1 mg),用体积分数为10%的甲醇水溶液溶解并转移至同一个100 mL 容量瓶中,定容,制得9 种甜味剂的混合标准储备液。
(2)标准工作溶液
分别准确移取0.05、0.125、0.5、1.25、2.5 和5 mL混合标准储备液,以及各0.5 mL 内标溶液,用水定容至10 mL 容量瓶中,配制得到6 级标准工作溶液。
1.2.2 样品处理与分析
准确称量0.5 g 电子烟烟液(精确至0.1 mg),转移至50 mL 具塞锥形瓶中,准确加入500µL 内标溶液和10 mL 水,然后于60%功率条件下超声萃取40 min,结束后摇匀,得萃取液。接着使用C18硅胶键合固相萃取柱对萃取液进行净化,即先加5 mL 甲醇对柱子进行活化,再加5 mL 水进行平衡,使柱子上筛板保留少部分水,然后移取1 mL 萃取液上样,弃去从柱子下筛板流下的溶液,再使用6 mL 体积分数为70%的甲醇水溶液进行淋洗,收集淋洗液。于70 ℃条件下将淋洗液氮吹浓缩至1 mL,进行HPLC-MS/MS 分析。分析条件:
色谱柱:Waters XBridge C18柱(150 mm×2.1 mm,填料粒径3.5 m);柱温:40 ℃;进样量:5µL;流速:0.2 mL/min;流动相:甲醇(A),5 mmol/L 乙酸铵的水溶液(B);梯度程序:0~15 min,A 由15%上升至90%,B 由85%下降至10%,15~22 min 为90%A和10%B,22~25 min 为15%A 和85%B;离子源:ESI;离子源温度:100 ℃;干燥气温度:300 ℃;干燥气流量:9 L/min;雾化气压力:275.79 kPa;毛细管电压:正离子4 000 V,负离子3 500 V;检测方式:多反应监测模式(MRM)。详细参数见表1。

表1 9 种甜味剂和内标的MRM 参数Tab.1 MRM parameters of nine sweeteners and internal standard
2 结果与讨论
2.1 质谱条件优化
取最高浓度混合标准溶液,在ESI 源正离子和负离子模式下进行质谱全扫描检测,得到被测化合物一级质谱图,找出每个化合物对应的母离子。然后进行二级质谱扫描,得到相应子离子。对每个化合物找出两对特征离子,分别进行裂解电压及碰撞能量优化,最后得到9 种甜味剂及内标的较佳质谱参数,见表1。结果显示,阿斯巴甜、阿力甜和纽甜同属二肽类化合物,在正离子模式下的响应较高,故采用[M+H]+作为母离子,而安赛蜜、糖精钠、甜蜜素、三氯蔗糖、华法林钠、新橙皮甙二氢查尔酮和甜菊糖苷在负离子模式下响应较好,其母离子依次选择为[M-K]-、[M-Na-2H2O]-、[M-Na]-、[M-2H]-、[M-H]-、[M-H]-和[M-H]-,其中三氯蔗糖一级质谱图中[M-2H]-(m/z 395)响应明显大于其他离子,故采用[M-2H]-为其母离子。此外,二级质谱图显示安赛蜜、甜蜜素、三氯蔗糖、新橙皮甙二氢查尔酮和甜菊糖苷等的碎片离子较少,故依次选择m/z 162、m/z 177、m/z 394.3、m/z 610.7 和m/z 802 为其对应的定性子离子。
2.2 色谱条件优选
为优选色谱条件,以加标样品进样溶液为对象,对比考察目标物在Waters XBridge C18色谱柱(150 mm×2.1 mm,5 m)、ZORBAX Eclipse XDB-C18色 谱 柱(150 mm×2.1 mm,3.5 m)和ZORBAX Eclipse XDB-C18色谱柱(150 mm×4.6 mm,3.5 m)上的保留分离情况,目标物出峰情况见图1a~图1c。可知,1#~10#色谱峰(后续图中编号顺序相同)依次为安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、阿力甜、华法林钠、新橙皮甙二氢查尔酮、纽甜和甜菊糖苷,目标物在3 根色谱柱上大都能被有效分离,其中4#三氯蔗糖和5#阿斯巴甜、7#华法林钠和8#新橙皮甙二氢查尔酮在前两根色谱柱上分别共流出,共流出目标物提取离子对色谱图显示能有效分离,而6#阿力甜、7#华法林钠和8#新橙皮甙二氢查尔酮在ZORBAX Eclipse XDB-C18色谱柱上共流出,共流出目标物提取离子对色谱图显示也能有效分离,且9#纽甜和10#甜菊糖苷出峰时间相比于前两根色谱柱发生了交替。综合考虑目标物整体出峰时间、响应大小及流动相消耗等情况,优选Waters XBridge C18色谱柱为分析用色谱柱。
进一步对比了甲醇-水、乙腈-水、甲醇-0.1%(体积比)甲酸水溶液、乙腈-0.1%(体积比)甲酸水溶液、甲醇-5 mmol/L 乙酸铵的水溶液和乙腈-5 mmol/L 乙酸铵的水溶液等不同流动相体系下目标物的色谱峰强度、峰形及分离效果。结果表明:乙腈为有机相时易导致个别组分共流出情况较甲醇严重,特别是极性较强的安赛蜜、糖精钠及甜蜜素峰形较宽、分离度较差。此外,在相同的梯度洗脱条件下,水相中加入甲酸后,安赛蜜、糖精钠及甜蜜素等有机盐的色谱峰形拖尾比较严重。总体比较,以甲醇-乙酸铵水溶液为流动相,所得到的色谱图峰形最好、响应强度较高,同时分离效果最好;另外为避免较高浓度的缓冲盐体系对质谱的影响,在满足要求的情况下选用5 mmol/L 乙酸铵缓冲盐体系。因此,最终确定甲醇-5 mmol/L 乙酸铵的水溶液为流动相。
在优化后的梯度洗脱条件下,混合标准溶液中9 种甜味剂以及内标的总离子流色谱图如图2所示。

图1 不同色谱柱条件下9 种甜味剂和内标的色谱峰Fig.1 Chromatogram peaks of nine sweeteners and internal standard using different chromatographic columns
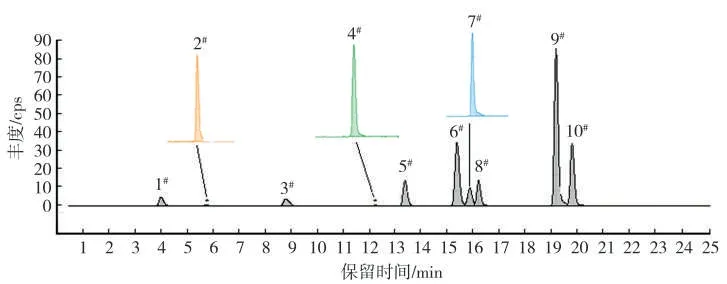
图2 优化梯度洗脱条件下9 种甜味剂和内标的色谱峰Fig.2 Chromatogram peaks of nine sweeteners and internal standard under optimized gradient elution conditions
2.3 固相萃取柱选择及淋洗条件优化
由于不同厂家烟液样品中乙醇、丙三醇等的质量分数不同,在进行液相色谱分离时这些醇类会对甜味剂在色谱柱中的保留性能产生影响。一般在分析甜味剂时,为了消减基质干扰,文献中常采用填料为C18、HLB 等固相萃取手段净化样品萃取液,以此来降低醇类等干扰物质对色谱峰形的影响,而且还可以减少离子化过程对目标物带来的干扰[10-11,21]。因为9 种甜味剂的分子结构、极性大小各不相同,在固相萃取柱的保留行为差异较大,在参考相关文献[6-7,21]的基础上,对填料为C18和HLB 的固相萃取柱进行了优选分析。取1 mL加标样品溶液进行上样和淋洗,收集各步骤的滤液,其中淋洗过程使用的溶剂按照洗脱强度从小到大的顺序,甲醇的体积分数依次为10%、20%、30%、40%、50%、60%、70%、80%、90%和100%的甲醇水溶液,用量分别为1 mL,采用优化好的仪器条件测定,考察各体积分数下目标物峰面积的变化情况,结果见图3 和图4。可以看出,就C18固相萃取柱而言,9 种目标物及内标在弃去的从柱子下筛板流下的溶液中均未检出,在淋洗液甲醇的体积分数为10%时,1#安赛蜜、2#糖精钠、3#甜蜜素被淋洗下来,且体积分数为50%左右时能被全部淋洗下来;体积分数增为20%时,4#三氯蔗糖、5#阿斯巴甜、6#阿力甜、7#华法林钠被淋洗下来,且体积分数为70%左右时能被全部淋洗下来;体积分数增至40%~50%时,8#新橙皮甙二氢查尔酮、9#纽甜和10#甜菊糖苷才被淋洗下来,且体积分数为90%左右时能被全部淋洗下来。相比而言,所有目标物在HLB 固相萃取柱的保留要更为强烈一些,主要原因是目标物在体积分数为20%时,1#安赛蜜、2#糖精钠、3#甜蜜素才被淋洗下来;8#新橙皮甙二氢查尔酮、9#纽甜和10#甜菊糖苷在体积分数为90%时在柱子上仍有部分保留,最后使用100%甲醇才被全部淋洗下来。综合考虑目标物在固相萃取柱上保留情况,基于保证各目标物回收率满足分析的同时尽可能去除萃取液中干扰组分的原则,本研究中选择C18固相萃取柱为净化柱子,以及体积分数70%的甲醇水溶液为淋洗液。
进一步优化淋洗液体积。取1 mL 加标样品溶液进行上样,丢弃柱子下筛板流下的溶液,再使用70%的甲醇水溶液进行淋洗,其中淋洗过程淋洗液使用体积分别为1、2、3、4、5、6、7 和8 mL,收集各体积条件下的滤液,浓缩至1 mL,采用优化好的仪器条件测定,考察不同淋洗液用量下目标物峰面积的变化情况,结果见图5。可以看出,使用2 mL 淋洗液,能将1#安赛蜜、2#糖精钠、3#甜蜜素、4#三氯蔗糖、5#阿斯巴甜全部洗脱下来。淋洗液体积增加至4 mL 左右时,能将保留更强的6#阿力甜、7#华法林钠和8#新橙皮甙二氢查尔酮全部洗脱下来。淋洗液体积增加至6 mL 时,9#纽甜和10#甜菊糖苷几乎100%被洗脱下来。因此,优选淋洗液体积为6 mL。
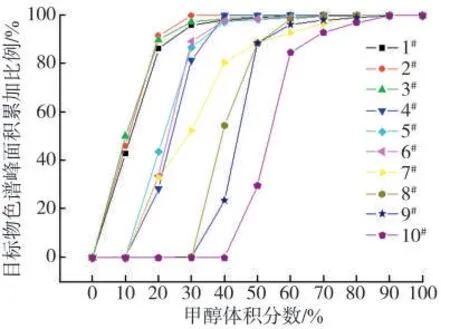
图3 C18固相萃取柱条件下淋洗液极性强度对9 种甜味剂和内标峰面积的影响Fig.3 Effects of eluent polarity on peak areas of nine sweeteners and internal standard using C18 SPE column
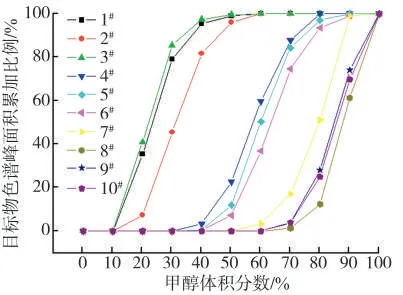
图4 HLB 固相萃取柱条件下淋洗液极性强度对9 种甜味剂和内标峰面积的影响Fig.4 Effects of eluent polarity on peak areas of nine sweeteners and internal standard using HLB SPE column
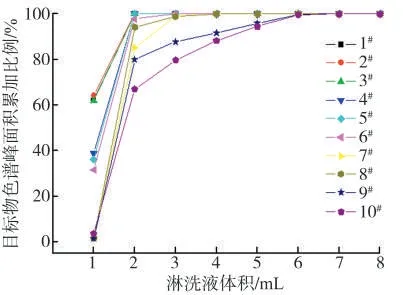
图5 不同淋洗液用量下9 种甜味剂和内标峰面积的变化情况Fig.5 Effects of eluent polarity on peak areas of nine sweeteners and internal standard under different eluent volume conditions
2.4 样品前处理条件优化
为优化样品萃取方式及水平条件,以实测结果为阳性的5#烟液样品(含有纽甜)为对象,考察振荡萃取和超声萃取方式下目标物质量分数的变化情况,结果见图6。可以看出,随萃取时间延长,目标物质量分数也逐渐升高,至40~50 min 时目标物萃取量基本达到最大值;整体上看,超声萃取结果略微大于振荡萃取结果。为此,在兼顾萃取结果与操作效率的基础上,优选样品萃取方式为超声萃取,萃取时间为40 min。
2.5 方法学验证
2.5.1 方法的线性范围、检出限及定量限

图6 不同萃取方式条件下目标物萃取量的变化情况Fig.6 Content of sweetener extracted by different extraction methods
采用优化的HPLC-MS/MS 条件对系列标准工作溶液进行测定,以系列标准工作溶液中各目标物浓度与内标浓度之比为横坐标,以对应的色谱峰面积之比为纵坐标,求得线性回归方程及其相关参数。以最低浓度标样为对象,平行测定9 次,求分析结果的标准偏差,分别以3 倍和10 倍标准偏差对应值为方法的检出限(LOD)和定量限(LOQ)。如表2 所示,9 种甜味剂在各自浓度范围内线性关系良好(回归方程的R2均大于0.998),9种甜味剂的检出限和定量限分别在0.005~0.287µg/mL 和0.017~0.956µg/mL 之间,可以满足各目标物定量分析要求。
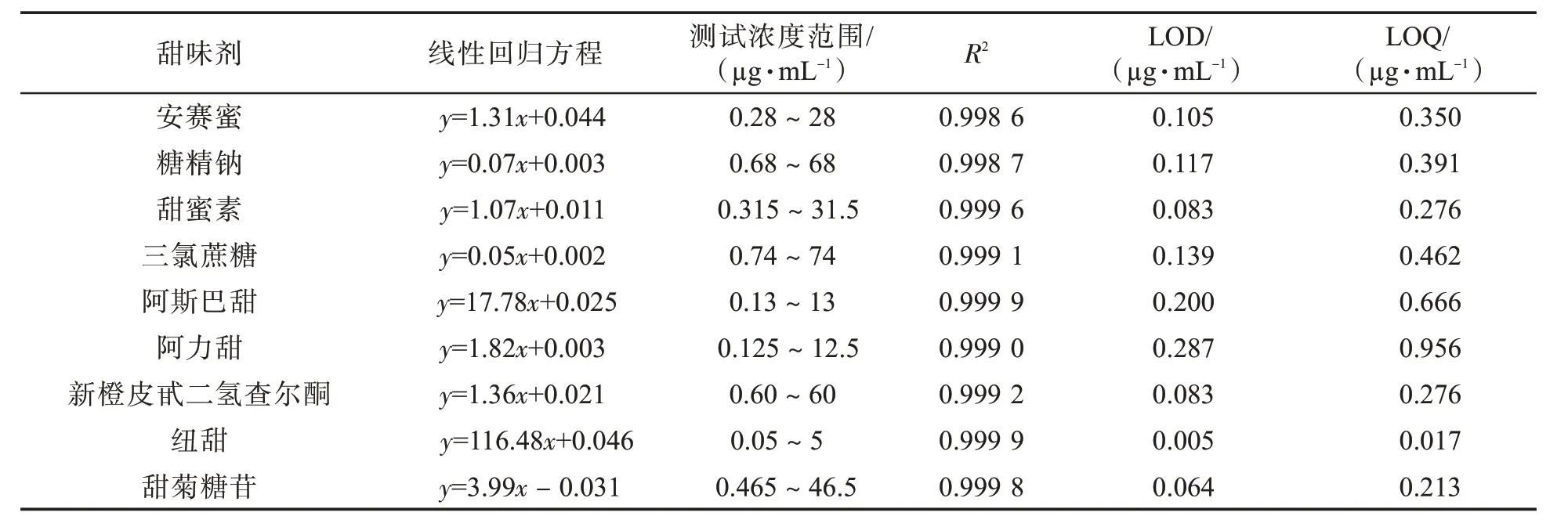
表2 9 种甜味剂的线性回归方程、测试浓度范围、R2、检出限及定量限Tab.2 Regression equations,range of concentrations,correlation coefficients,limits of detection(LODs)and limits of quantification(LOQs)of nine sweeteners
2.5.2 方法的准确度和精密度
参照相关标准检测方法确认的回收率和精密度测试要求[22],以样品进样溶液为对象,通过加标样品的回收率实验来验证方法的准确度,并以RSD 来评价精密度。在低、中、高3 个不同浓度水平(分别为2、10 和20 倍LOQ)下进行回收率实验,每个浓度水平重复测定6 次,计算样品中各种甜味剂的回收率和RSD,结果见表3。可知,9 种甜味剂在不同浓度水平下的加标回收率在82.96%~114.01%之间,RSDs 在2.19%~5.18%之间,表明方法具有较好的准确度和重复性,能够满足微量目标物的定量分析。

表3 不同浓度下9 种甜味剂的加标回收率和相对标准偏差(n=6)Tab.3 Spiked recoveries and RSDs of nine sweeteners at different concentrations(n=6) (%)
2.6 实际样品的测定
采用本方法,对市售8 种不同品牌的电子烟烟液进行3 次平行测定,结果(表4)显示在1 个样品中检出纽甜,质量分数为5.92 mg/kg,且相对标准偏差<5%。总的看来,检测结果符合GB 2760—2014《食品安全国家标准 食品添加剂使用标准》中有关甜味剂的使用限量要求。
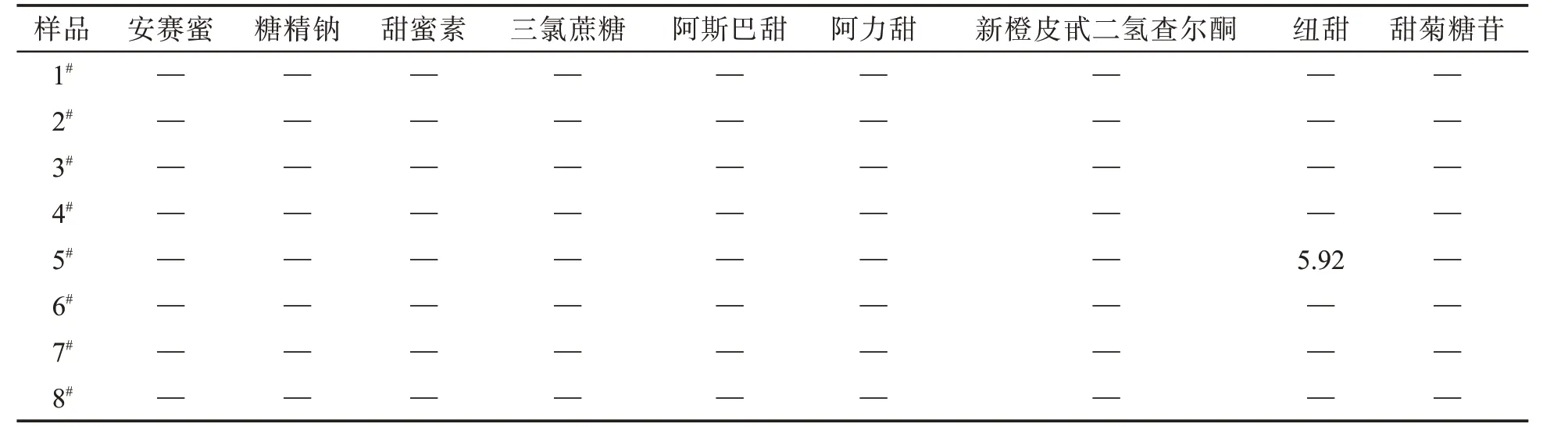
表4 实际样品的测定结果Tab.4 Test results of commercial samples (mg·kg-1)
3 结论
①通过优化目标物质谱和色谱条件,以及优化样品净化和提取方式,建立了SPE-HPLC/MS/MS 法同时测定电子烟烟液中安赛蜜、糖精钠、甜蜜素、三氯蔗糖、阿斯巴甜、阿力甜、新橙皮甙二氢查尔酮、纽甜、甜菊糖苷等的方法。②在优化条件下,目标物的检出限和定量限分别介于0.005~0.287µg/mL 和0.017~0.956µg/mL 之间,加标回收率在82.96%~114.01%之间,RSDs 在2.19%~5.18%之间。③实际样品检测结果表明,定量结果准确可靠,可为电子烟烟液中甜味剂的筛查和检测提供方法参考。
