Carvajal综合征一例伴桥粒斑蛋白基因新突变
2020-06-05吴维郑璐瑶潘超兰杨挺李明
吴维 郑璐瑶 潘超兰 杨挺 李明
1无锡市儿童医院皮肤科214023;2上海交通大学医学院附属新华医院皮肤科200092
通信作者:李明,Email:liming01@xinhuamed.com.cn
Carvajal综合征是一种罕见的遗传性心脏皮肤综合征,其致病基因为桥粒斑蛋白(desmoplakin,DSP)基因,临床表现为羊毛状发、掌跖角化和扩张型心肌病等[1⁃2],也可伴发白甲病、牙齿异常、听力减退及反复感染[3⁃5]。该病的症状通常随着时间的推移而显现[6]。羊毛状发可出生既有,掌跖角化一般在婴儿期显现,心脏症状早期不一定出现[5⁃6]。我们报道1 例表现为羊毛状发及掌跖角化但无心脏症状的Carvajal综合征,并对其进行基因检测。
病历资料
患儿女,3 岁。出生时头发卷曲,外观呈羊毛状,8 月龄时掌跖部出现点状角化过度,渐累及膝盖,无胸痛、心悸、呼吸短促,心电图、心脏彩超检查均未见异常。父母体格检查和心脏彩超检查均未见异常,非近亲结婚,家族中无类似病例。体检:一般情况可,身高、体重和智力均在正常范围。头颅无畸形,心肺肝脾未见异常,四肢肌张力正常,神经系统检查无特殊。皮肤科检查(图1):头发卷曲,稍稀疏,呈羊毛状改变,眉毛、睫毛未见异常。掌跖轻度点状角化过度,膝盖角化性丘疹,余皮肤未见异常。患儿无牙齿、黏膜、指甲、汗腺等其他外胚层发育异常表现。
基因突变检测
1. 外周血基因组DNA 提取:经上海交通大学医学院附属新华医院医学伦理委员会批准(批件号XHEC⁃D⁃2019⁃115),并经患儿父母签署知情同意书后,分别采集患儿及其父母外周血2 ml。应用QIAamp DNA Blood MinikitⅠ(德国QIAGEN 公司)试剂盒提取全血基因组DNA。同样方法提取100例无亲缘关系的健康个体血样基因组DNA 作为对照。
2.二代靶向测序和Sanger测序:使用上海安百隆生物科技有限公司设计的遗传性皮肤病检测专用Panel(由罗氏公司合成),用IlluminaHiseq X Ten高通量测序仪测序,在相关变异频率数据库[寡核苷酸多态性数据库(dbSNP)、千人基因组数据库、ClinVar数据库和人类基因突变数据库(HGMD)]中对致病变异位点进行评估,根据美国医学遗传学与基因组学学会(ACMG)遗传变异分类指南及患者的临床表型进行致病变异的筛选。使用Primer⁃BLAST 工具(https://www.ncbi.nlm.nih.gov/)设计基因 引 物(DSP ⁃ E23:正 向 引 物 序 列 5′ ⁃AGAGGACTGTGAAGGACCAG⁃3′和反向引物5′⁃AAGAACAGCAGGGCACACAG⁃3′;DSP⁃E24:正向引物5′⁃TTTGATGGGCTGAGGAAGAA⁃3′和反向引物5′⁃ATTGACAGTGGACGGTCTCA⁃3′),并通过PCR 扩增,用ABI3730XL 型全自动测序仪进行Sanger测序。
3.测序结果:见图2。患儿DSP基因第23号外显子发生移码突变c.5152dupT(p.L1718Ffs*15),即编码区第5152 位核苷酸T 重复,导致编码蛋白第1718 位亮氨酸变为苯丙氨酸,随后第15 位氨基酸出现1个终止氨基酸;还检测到第24号外显子存在无义突变c.C6478T(p.R2160X),即编码区第6478位核苷酸由C变为T,导致编码蛋白第2160位精氨酸变成终止氨基酸。患儿母亲第23号外显子亦检测到移码突变c.5152dupT(p.L1718Ffs*15),而其第24 号外显子未检测到基因突变。患儿父亲、100例健康对照中均未检测到以上2个DSP基因突变。
诊断与治疗
根据患儿临床表现和DSP基因检测结果等,诊断为Carvajal 综合征。患儿掌跖角化相对轻微,目前未特殊处理;每半年进行心脏全面检查(心电图、动态心电图监护、心脏超声等),目前未发现异常。
讨 论
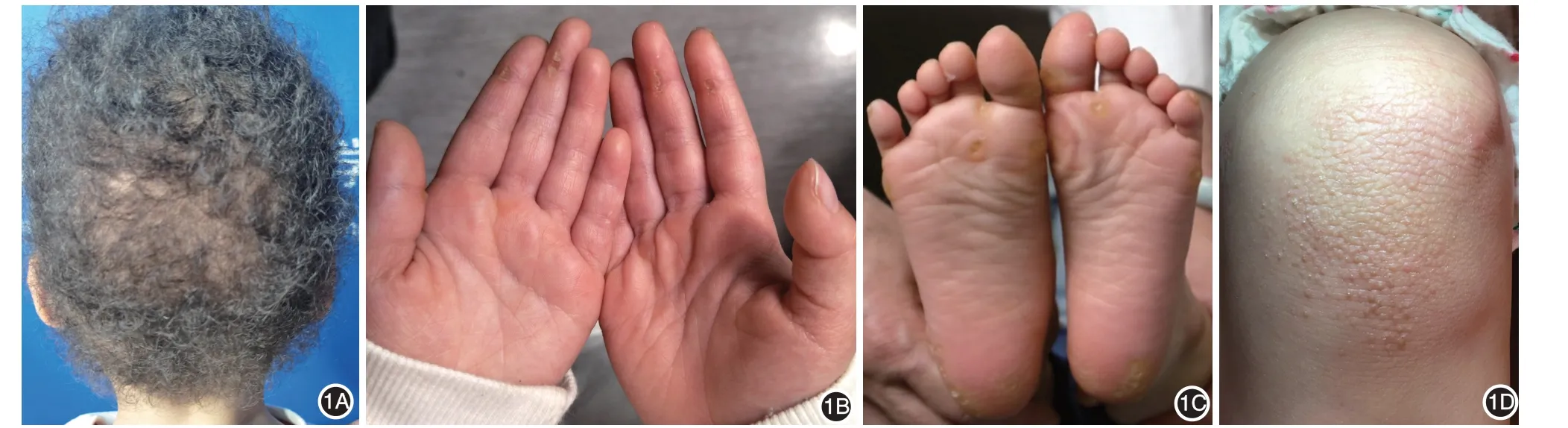
图1 患儿3岁时,头发卷曲,稍稀疏,呈羊毛状改变(1A);掌跖轻度点状角化过度(1B、1C);膝关节伸侧角化性丘疹(1D)

图2 患儿、其父母及健康对照桥粒斑蛋白(DSP)基因突变分析 患儿DSP基因第23号外显子发生移码突变c.5152dupT,第24号外显子发生无义突变c.C6478T;患儿母亲DSP基因第23号外显子存在移码突变c.5152dupT,第24 号外显子未发现突变;患儿父亲及健康对照第23、24 号外显子相应位点均未发现突变
Carvajal综合征是一种罕见的遗传性心脏皮肤综合征,目前国内外报道不多,其中国内中文报道1 例[7]。Carvajal 综合征可认为是另一种心脏皮肤综合征Naxos病的一种变异型[8],Naxos病也表现为羊毛状发、掌跖角化,但心脏表现主要为心律失常性右心室心肌病,致病基因为斑珠蛋白[9]。Carvajal综合征主要为常染色体隐性遗传,突变热点区域主要在DSP基因第23、24号外显子,少数为常染色体显性遗传,主要表型除羊毛状发、掌跖角化和扩张性心肌病外,可伴有牙齿异常,突变区域主要在DSP 基因第13、14 号外显子[10]。经二代靶向测序分析,我们发现本例患儿的突变基因为DSP 基因。DSP是桥粒形成过程中的一个锚定蛋白,直接或间接与其他几个形成桥粒的蛋白相互作用。DSP 基因大致可分为N 端、Rod 区域和C 端3 个主要区域[11],3个区域内都有引起Carvajal 综合征、皮肤脆性及严重棘层松解性大疱性表皮松解症的突变位点,位于N端的p.Q331*突变可只引起掌跖角化,而C 端p.R2522Sfs*39 和p.R2586*突变的患儿仅毛发和皮肤异常,而无心脏病变[10]。因此,基因型与表型的相关性不易预测[10]。
本例患儿携带复合杂合突变,分别为DSP基因第23 号外显子移码突变c.5152dupT(p.L1718Ffs*15)和第24号外显子无义突变c.C6478T(p.R2160X),前者来自于其母亲。患儿表型高度符合Carvajal综合征,且患儿父亲及100例健康对照中均未检测到上述2个DSP基因突变,此2种突变在东亚人群中的突变频率为0,提示为致病突变,而非单核苷酸多态性。检索HGMD 数据库(http://www.hgmd.org)及相关文献,未见报道移码突变c.5152dupT(p.L1718Ffs*15)。此突变位于DSP 基因Rod 区域(c.3585⁃5379或p.1195⁃1793),该片段仅存在于DSP 异构体Ⅰ(DSPⅠ)中,另外2 种异构体分别为DSPⅡ和DSPⅠa,它们缺少Rod 区中央α-螺旋杆状结构域的一部分,DSPⅡ缺少氨基酸1195-1793,DSPⅠa缺少氨基酸1351- 1793[10,12]。DSPⅡ是调节角质形成细胞黏附的主要异构体[13],皮肤基底层桥粒蛋白的错位可间接反映心肌细胞间闰盘的情况[14]。对一DSP 基因突变位点为p.E1493X(位于DSPⅠ)心律失常性心肌病家系研究[15]发现,7例突变携带者中仅1例表现出卷发及皮肤异常,且心脏症状普遍轻微,皮肤免疫组化染色显示,DSP 和斑珠蛋白的分布与对照皮肤相当。目前,本例患儿亦未发现心脏异常表现,其母亲是无临床症状的c.5152dupT基因突变携带者,符合上述文献的观点。
本例患儿DSP 基因还存在无义突变c.C6478T(p.R2160X),其父母及100例健康对照者均未检测到该突变,另外父亲的口腔黏膜脱落细胞、毛发、精液中也未发现此突变,提示其为新生突变。此突变位于DSP基因的C末端,是与角蛋白中间丝相互作用的关键部位,p.R2522Sfs*39 和p.R2586*突变的患儿仅出现毛发和皮肤异常,无心脏病变[10]。同样发生在DSP 基因C末端p.G2375A 和p.L2329P 突变的兄妹[6],父母亦无临床表现,其中先证者男,11岁,表现为局灶性掌跖角化、羊毛状发和扩张性心肌病,其妹5 岁且有相似的临床表现,但无心脏累及,其妹4岁后每6个月进行全面的心脏检查,无任何异常表现。目前,p.R2160X突变在Carvajal 综合征中未见文献报道,但在心律失常性右心室发育不良及心律失常性右心室心肌病中为致病性突变[16⁃17]。目前,仍无法准确预测本例患儿的预后,需要定期随访并进行心脏全面检查,早期发现并进行治疗。此外,本例出现的2种突变也存在无义介导mRNA降解的可能[18]。
利益冲突 所有作者均声明不存在利益冲突
