当归-白芍药对HPLC 指纹图谱建立
2020-06-02闫宏丽马旭彤邓秀平宋纹苗淑杰刘志东
闫宏丽 马旭彤 邓秀平宋 纹苗淑杰刘志东∗
(1.天津中医药大学,现代中药发现与制剂技术教育部工程中心,天津301617; 2.天津中医药大学,天津市现代中药重点实验室-省部共建国家重点实验室培育基地,天津301617; 3.天津同仁堂集团股份有限公司,天津300385; 4.天津宏仁堂药业有限公司,天津300122)
药对又称对药,最早见于《黄帝内经》 中的半夏秫米汤治疗胃不和则卧不安症[1],是2 味中药固定的配伍形式,把中药和方剂巧妙的联系在一起[2]。药对配伍的主要依据是中医基础理论,即四气五味、升降沉浮、归经及有毒无毒[3]。药对的组成虽简单,但可较全面的兼顾病情,最大限度发挥治疗作用,体现适证化裁、灵活加减的优点[4]。
当归-白芍为临床常用的养血理血药对,来源于《金匮要略》 之“当归芍药散”[5]。且多与其他药物配伍成别方应用于临床,如《四物汤》 《逍遥散》 《温经汤》 《大秦艽汤》 《当归芍药散》 等。当归释名干归、山蕲、白蕲、文无,为伞形科植物当归Angelica sinensis(Oliv.) Diels 的干燥根,归心、肝、脾经,具有补血活血、调经止血、润肠通便的功效。白芍来源为毛莨科植物芍药Paeonia lactifloraPall 的干燥根,性微寒,味苦、酸。归肝经、脾经,具有补血敛阴、平肝止痛的功效[6]。辛香之当归(血中气药)与白芍阴柔补血之品(血中血药) 相配使用,二者相须相使[7],补血不滞血、温而不燥,可用于补血调血。在现代药理研究表明,当归中藁本内酯、阿魏酸等可抑制血小板聚集,增强巨噬细胞的吞噬功能,延长血栓的形成,具有抗血栓、提高机体免疫力的作用[8-10];白芍中白芍总苷具有抗炎、镇痛、保肝、抗氧化的作用[11-12],没食子酸等可起到抑菌杀菌的作用[13-15]。因此,当归-白芍药对配伍使用,可起到很好的养血调肝、健脾利湿功效[16-17]。
当药物的物质基础不明确时,指纹图谱可全面地反映复方中所含化学成分的种类和数量。不同产地的当归、白芍其活性成分含有量有差别,单味当归、白芍及含当归、白芍中药复方的指纹图谱已有报道[18-21],但关于当归-白芍配伍药对的指纹图谱研究尚未见报道。本实验建立当归-白芍药对HPLC 指纹图谱,以期为中药配伍规律研究、临床用药以及中成药研制提供参考。
1 材料
1.1 仪器 Agilent 1260 高效液相色谱仪(配置G1314B VWD 检测器,美国安捷伦公司);JA2103N 天平(千分之一,上海民侨精密科学仪器有限公司);FA124 天平(万分之一,天津亿诺科学仪器有限公司);KQ-400B 高功率数控超声波清洗器(昆山市超声仪器有限公司);N-1100 旋转蒸发仪(上海爱朗仪器有限公司);ZDHW 调温电热套(北京中兴伟业仪器有限公司);FDU-1200 真空冷冻干燥机(上海爱朗 仪器有 限公司);去离子水机 (美 国Millipore 公司)。
1.2 试剂与药物 对照品没食子酸(批号10831-201605,纯 度 90.80%)、芍药苷 (批 号 110736-201842,纯 度97.40%)、阿魏酸(批号110773-201614,纯度99.00%),均购自中国食品药品检定研究院;对照品芍药内酯苷(批号Y21A9H59553,纯度91.40%)、苯甲酰芍药苷 (批号P21A9S68357,纯度98.00%) 均购自上海源叶生物科技有限公司。甲醇(分析纯,天津市康科德科技有限公司);乙腈(色谱纯,美国Fisher 公司);磷酸(色谱纯,美国赛默飞世尔科技有限公司);去离子水由Milli-Q 超纯水系统过滤得到。
不同产地当归、白芍药材,经天津宏仁堂药业有限公司宋纹高级工程师和天津同仁堂集团股份有限公司苗淑杰教授级工程师鉴定为正品,信息见表1。

表1 样品信息
2 方法与结果
2.1 对照品溶液制备 精密称取没食子酸、芍药内酯苷、芍药苷、阿魏酸、苯甲酰芍药苷对照品适量,置于10 mL量瓶中,加甲醇定容,制得各对照品质量浓度分别为68.3、52.8、41.2、54.6、53.5 μg/mL 的混合对照品溶液,4 ℃保存,备用。
2.2 煎液制备 当归-白芍合煎液,精密称取当归、白芍各10 g,混合,置500 mL 圆底烧瓶内,加8 倍量去离子水,浸泡30 min,用调温电热套以220 V 加热至沸腾后调至75 V 保持微沸,回流提取1 h,两层纱布滤过;药渣用8 倍量的去离子水再次回流提取1 h,两层纱布过滤,合并2 次提取液,减压浓缩至稠膏状,置于-45 ℃冷旋预冻4 h后,置于-80 ℃的冷冻干燥仪中干燥24 h,得浅黄褐色当归-白芍干膏粉(无明显气味,易吸潮),出膏率(23.45±2.53)%,待用。
称取制得的各个产地干膏粉0.15 g,于50 mL 三角瓶中,精密移取60%甲醇10 mL,摇匀,分别超声(400 W、50 kHz) 处理30 min,取出室温冷却后,60% 甲醇补足减失质量,摇匀,室温静置30 min,取上清液,过0.22 μm有机微孔滤膜,即得。分别吸取10 μL 进液相色谱仪。
2.3 色谱条件 Waters X Bridge Shield RP18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈(A) -0.1% 磷酸水(B),梯度洗 脱 (0 min,5% A;10 min,22% A;25 min,32%A;30 min,75% A;33 min,5% A;40 min,5%A);体积流量0.8 mL/min;柱温35 ℃;检测波长230 nm;进样量10 μL。
2.4 方法学考察
2.4.1 精密度试验 取当归-白芍药对,按“2.2” 项下方法制备供试品溶液,在“2.3” 项色谱条件下,连续进样6次,测得各共峰相对保留时间RSD 0.03%~0.29%,各共有峰相对峰面积RSD 0.41%~1.15%,表明仪器精密度良好。
2.4.2 重复性试验 取当归-白芍药对,按“2.2” 项下方法平行制备供试品溶液6 份,在“2.3” 项色谱条件下,分别进行测定,测得各共有峰相对保留时间RSD 为0.02%~0.16%,各共有峰相对峰面积RSD 为0.33%~4.19%,表明该方法重复性良好。
2.4.3 稳定性试验 取当归-白芍药对,按“2.2” 项下方法制备供试品溶液,在“2.3” 项色谱条件下,分别于0、6、12、24、36、48、72 h 依次进样,测得各共有峰相对保留时间RSD 0.55%~0.87%,各共有峰相对峰面积RSD 0.79%~4.95%,表明供试品溶液在72 h 内稳定性良好。
2.5 指纹图谱建立及评价
2.5.1 不同产地当归-白芍药对指纹图谱 根据各色谱峰的相对保留时间,确定了16 个共有峰并指认出5 个特征峰,分别为没食子酸、芍药内酯苷、芍药苷、阿魏酸、苯甲酰芍药苷。建立了当归-白芍药对的对照指纹图谱,见图1;按“2.2” 项下方法分别制备18 批供试品溶液,在“2.3” 项色谱条件下依次进样,相似度见表2;HPLC 指纹图谱见图2。表2 显示,不同产地18 批当归-白芍药对相似度较高,大于0.99,故不同产地18 批当归-白芍药对整体化学组分相似,制备工艺稳定,但化学成分的含有量可能存在差异。

表2 18 批样品相似度
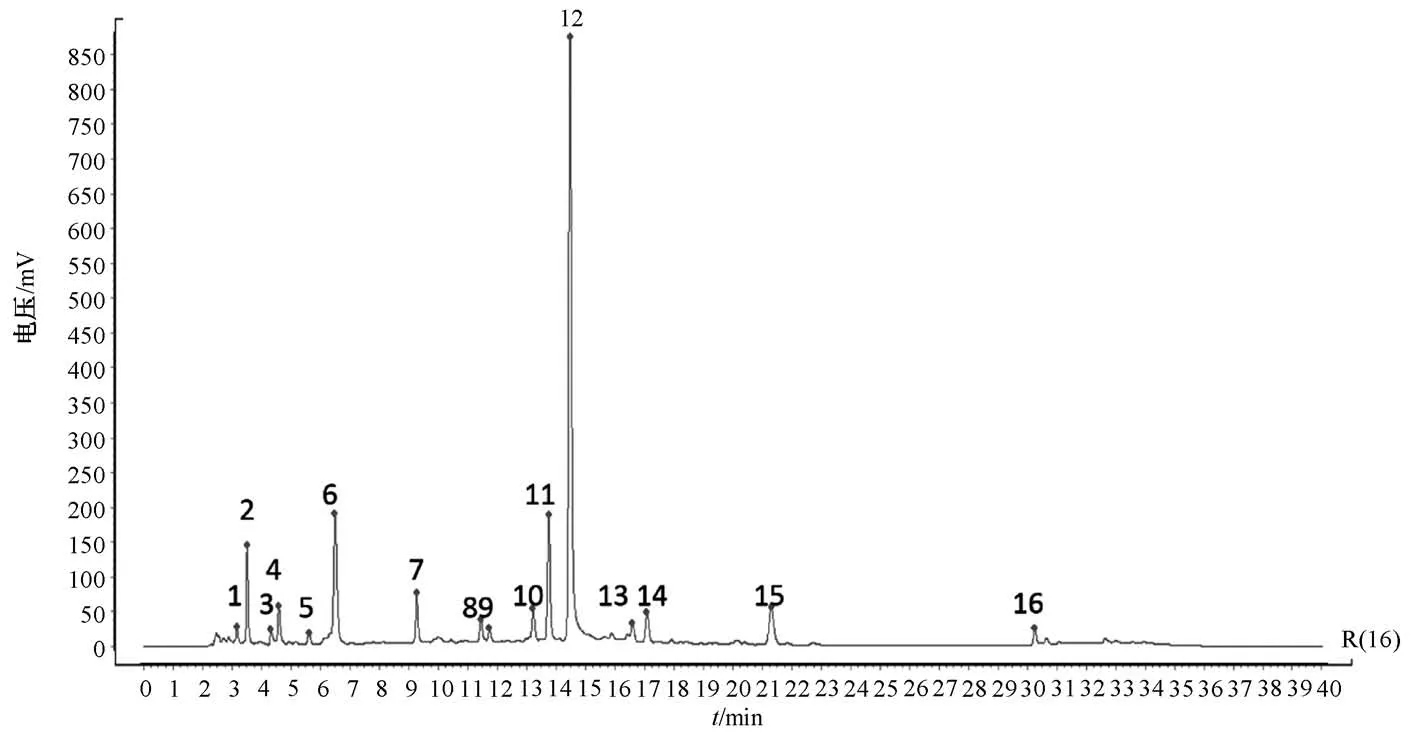
图1 当归-白芍样品对照指纹色谱图

图2 不同产地18 批样品HPLC 指纹图谱
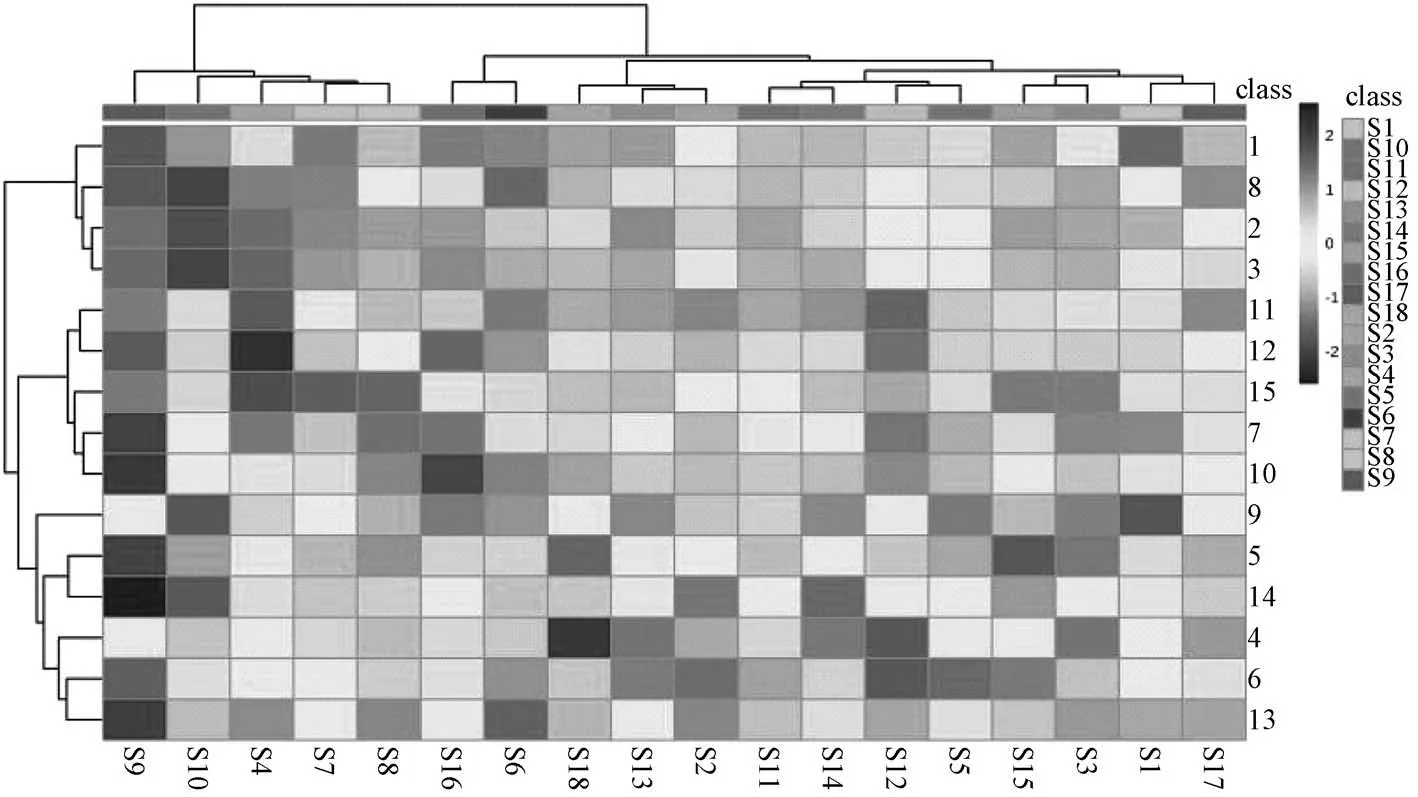
图3 不同产地18 批样品聚类分析图
2.5.2 聚类分析 使用Metabo Analyst,对18 批样品中16个共有峰峰面积标准化后进行聚类分析,见图3。18 批样品可被分为2 大类,S4、S7、S8、S9、S10 为一类,其余为一类。即甘肃岷县的当归和山东的白芍为一类,其余为一类。由此表明,甘肃岷县的当归和山东荷泽的白芍该组合的指标成分质量相近,与其他产地的当归-白芍可明显划分,故认为不同产地的药材之间可能由于种植的土壤、温度、光照和水分等不同而产生差异。
3 讨论
3.1 色谱条件 当归-白芍药对多以汤剂用于临床,为保证与传统煎剂尽可能相符,且活性成分极性较大,故以去离子水作为提取溶剂。经文献调研及经方中当归-白芍药对的用药比例,确定当归-白芍药对的配伍比例为1 ∶1[22-23]。本实验比较甲醇-水、乙腈-水洗脱系统,由于甲醇在低波长区吸收明显,采用最大吸收法产生的基线有波动;在水相中加入磷酸后,阿魏酸和芍药苷峰形较理想,且各个峰分离效果好、基线平稳。故选择乙腈-磷酸水(0.1%) 为流动 相。对体积流量 (0.7、0.8、0.9 mL/min)、柱温(30、35、40 ℃) 进行考察。0.9 mL/min 较0.8 mL/min 峰型较拥挤,0.7 mL/min 与0.8 mL/min 差别不大,为缩短分析时间,故选择0.8 mL/min 作为最佳体积流量。在3 种柱温下色谱峰无明显差异,为使色谱条件更耐用,选择35 ℃。对检测波长(222、230、254、275 nm) 进行考察,本研究前期使用DAD 检测器全波长扫描,在230 nm 波长下基线平稳,共有峰数量多,各个峰吸收较大、干扰较少,且分离度符合检测要求,部分峰具有较高响应值,结合2015 年版《中国药典》 最终选择230 nm 波长进行后续检测。
3.2 样品制备 为了方便样品的保存及检测到更多成分,本实验将煎液进行浓缩富集。根据当归-白芍药对中主要化学成分的溶解性、极性等性质,本研究选择25%甲醇、乙醇、乙腈;50% 甲醇、乙醇、乙腈;75% 甲醇、乙醇、乙腈及纯水作为当归-白芍药对干膏粉的复溶溶剂,结果显示25%甲醇、乙醇、乙腈,75%乙醇、乙腈超声后粉末仍未全部溶解,粉末呈棕黄色;50%乙腈、纯水作为复溶溶剂时,各指标成分不能充分提取;故细致考察后,定60%甲醇为复溶溶剂。供试品溶液制备的超声时间和功率对当归-白芍药对的各共有峰峰型无明显影响,为提高提取效率保证有效成分完全提取,故选超声及功率分别为30 min、400 W。研究还对当归-白芍药对浸膏的干燥方式(真空干燥、喷雾干燥及冷冻干燥) 进行考察,结果可知,冷冻干燥后各指标成分较为稳定,且样品损失率低,故选择冷冻干燥。
不同产地18 批样品与对照指纹图谱的相似度均大于0.99,表明不同产地的当归-白芍药对间相似度良好。但单一的相似度评价难以全面地评价不同产地当归-白芍药对的差异性。为了更全面、整体地分析不同产地当归-白芍药对的质量差异,又采用化学模式识别系统中聚类分析进行分析[24-25],可明显区分不同产地配伍药对的质量。
本研究建立了当归-白芍药对HPLC 指纹图谱,对部分共有峰进行了化学成分指认。该方法简便、快捷、准确且灵敏度高,能够较全面地反映当归-白芍药对的化学成分信息,可用于其全面质量控制。
