利用高通量测序技术探究肠球菌属在Ⅲ期与Ⅳ期结肠癌患者间的丰度差异
2020-06-01王玉柳明张巍远张宇坤胡汉卿黄睿汤庆超陈瑛罡王贵玉
王玉柳明 张巍远 张宇坤 胡汉卿 黄睿 汤庆超 陈瑛罡 王贵玉
近年来,结直肠癌(colorectal cancer,CRC)的发病率和死亡率逐年攀升,成为威胁国民健康的重大疾病[1]。针对结直肠癌的预防、诊断和治疗的研究日渐增多,尤其是对于分子机制及治疗靶点的研究已成为热点内容。肠道菌群与结直肠疾病有着密切关联,如具核梭杆菌、大肠杆菌和粪肠球菌等已被证明在结直肠癌的发生和发展中发挥重要作用,可以作为未来结直肠癌防治的潜在靶点[2-6]。随着高通量测序技术的日趋成熟,利用粪便样本研究结直肠疾病患者的肠道菌群特征成为便捷、可靠的实验手段。目前的研究表明,肠球菌属(Enterococcus)可以发挥“抑瘤”作用,高丰度的肠球菌属可能降低结直肠癌的发病风险[6]。但亦有研究表明,肠球菌属可能通过诱导胞外过氧化物介导DNA 损伤等机制发挥“促瘤”作用[7]。
进展期结肠癌是结肠癌中的特殊类型,其治疗、预后及菌群特征均不同于非进展期结肠癌[8]。而其中Ⅲ期与Ⅳ期结肠癌患者的菌群特征差异尚缺乏充足的研究,肠球菌属作为“明星”菌属在两组间的丰度差异仍不明确。因此,基于高通量测序技术探究Ⅲ期与Ⅳ期结肠癌患者肠球菌属丰度差异不仅可以明确Ⅲ期与Ⅳ期结肠癌患者的菌群特征差异,更将有助于后续的基础研究及为临床转化提供目标和方向。
材料与方法
一、样本采集及入组标准
收集2018 年7 月至2019 年1 月,于哈尔滨医科大学附属第二医院结直肠肿瘤外科就诊的结肠癌患者粪便样本。所有样本均于患者入院当天未进行医疗处置时采集。本研究的入组标准如下:(1)患者通过病理活检及影像学评估确认为Ⅲ期结肠癌或结肠癌肝转移(Ⅳ期);(2)患者于本科室行原发灶切除手术。本研究的排除标准如下:(1)样本采集前3 个月内服用抗生素或皮质类固醇激素或益生菌治疗者;(2)样本采集前3 个月内接受过腹部手术或其他侵入性治疗者;(3)样本采集前1 周内服用导泻剂或行灌肠治疗或行肠镜检查者;(4)有癌症个人史、家族史或炎症性肠病个人史者;(5)特殊饮食者[2,8];(6)术前进行放疗或新辅助化疗者;(7)既往进行过粪菌移植治疗者;(8)信息不完善或未取得知情同意者。根据入组标准和排除标准,共收集符合标准的Ⅲ期结肠癌患者粪便样本17例,结肠癌肝转移(Ⅳ期)患者粪便样本10 例。获取粪便样本后立即放入-80 ℃冰箱保存备用。所有进行粪便样本采集的患者均已取得其同意,并签署知情同意书。本研究已经被哈尔滨医科大学附属第二医院伦理审查委员会批准进行。
二、倾向值匹配法
为排除入组患者基线资料不一致导致的菌群分析结果差异,我们采用倾向值匹配法(PSM)均衡组间基线资料。运用Logistic 回归模型分析27 例患者的基线资料变量赋值,所得P值即为倾向值,按最近匹配法1:1 配对,卡钳值取0.1。我们选取患者的性别、年龄、身体质量指数(BMI)、美国麻醉师协会(ASA)评分和肿瘤部位作为变量。其中,性别、年龄、BMI、肿瘤部位是患者的基本特征。ASA 评分是一个简单、主观的量化标准用以评估患者的身体状况及手术风险,在临床工作和研究中应用广泛[9]。通过倾向值匹配法,纳入基线资料基本一致的9 例Ⅲ期患者和9 例Ⅳ期患者入组,对其粪便样本进行高通量测序和生物信息学分析。
三、实验方法
1.DNA 提取:粪便样本微生物总DNA 的提取采用德国QIAGEN 公司生产的QIAamp DNA Stool Mini Kit 试剂盒。具体步骤按照说明书操作,所提取的DNA 于-80℃冰箱保存[10]。
2.高通量测序:高通量测序扩增细菌16S rRNA的338F~806R 区域,上下游引物序列分别为338F 5'-ACTCCTACGGGAGGCAGCAG-3',806R 5'-GGACTACHVGGGTWTCTAAT-3'。将得到的PCR产物构建测序文库,采用Illumina Miseq 平台测序,对高质量测序数据进行生物信息学分析,该部分由上海美吉生物医药科技有限公司完成。
四、统计学分析
采用统计学软件SPSS 22.0 进行倾向值匹配法和统计学分析。组间差异采用方差分析,符合正态分布、方差齐性的数据采用t检验,不满足正态分布或者方差齐性的数据采用Wilcoxon 秩和检验,以P<0.05 认为差异有统计学意义。基于样品测序产生的OTU(Operational Taxonomic Units)结果,采用Qiime 软件计算beta 多样性距离矩阵,并用R 语言vegan 软件包作非线性多维标度(nonmetric multidimensional scaling,NMDS)[10]。
结果
一、倾向值匹配法结果及匹配后病理信息
选取性别、年龄、BMI、ASA 评分和肿瘤部位作为变量进行倾向值匹配,匹配前及匹配后两组基线信息详见表1。此外,我们对匹配后的两组除综合病理分期之外的其他病理信息进行了比较(见表2)。总体来看,经过倾向值匹配后,Ⅲ期组和Ⅳ期组除综合病理分期不同外,在基线资料和病理信息上差异没有统计学意义,这便于我们进行后续的测序数据分析比较。
二、测序基本信息
总计18 例样本进行了测序分析,共获得原始序列1 552 674 条。经过质量过滤和去除嵌合体之后的优质序列有1 044 208 条,样本序列平均长度主要集中在421~441 bp 区域。为了便于进一步研究,对于所有有效序列进行了OTU 聚类分析,按照最小样本序列数进行了抽平,最终获得了283 个OTU。以测序数据量为横坐标,以香农指数为纵坐标,作Shannon-wiener 曲线图(见图1)。从图中可以看出,曲线在开始时迅速上升,说明随测序深度增加,新发现的菌种在逐渐增多。随着序列数增多(测序深度增加),曲线逐渐趋于平缓,进入平台期,说明测序深度达到要求,覆盖了所有菌种。

表1 倾向值匹配结果[例(%)]

表2 匹配后患者病理信息表[例(%)]
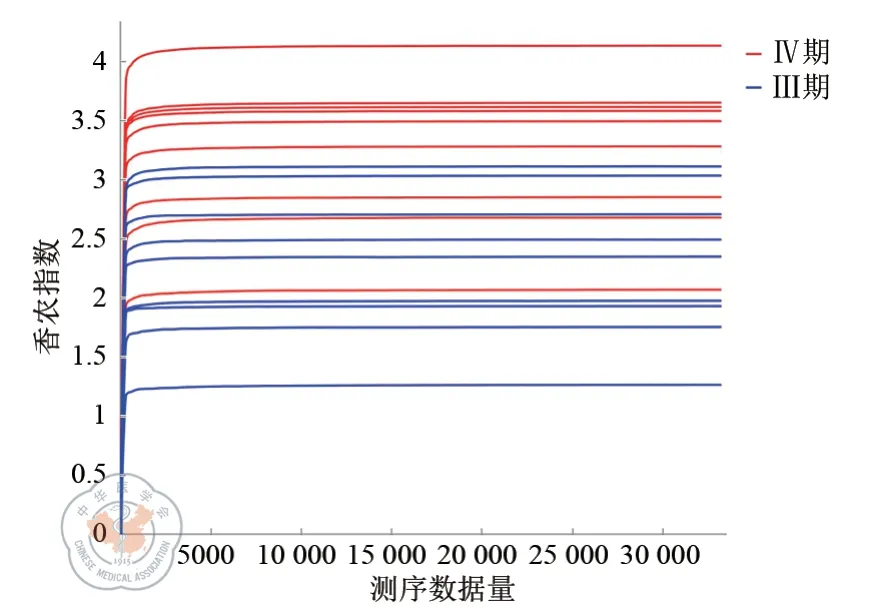
图1 Shannon-wiener 曲线图
三、Alpha 多样性分析及群落组成分析
Alpha 多样性分析是反映样本物种组成丰富度和多样性的重要指标。对两组样本进行基于OTU 水平的Alpha 多样性差异分析,可以有效反映两组在物种组成丰富度和多样性层面的差异。我们选取了sobs 指数、chao 指数、ace 指数来反映物种丰富度,采用辛普森(simpson)指数、香农(shannon)指数来反映物种多样性。指数组间差异性检验提示,Ⅲ期组与Ⅳ期组在物种多样性和丰富度上差异有统计学意义(P<0.05)(见表3)。此外,我们在属水平进行了群落组成分析,选取丰度前15 的菌属绘制了Heatmap 图(见图2)。群落组成分析结果提示,两组在菌属水平的群落组成上有着明显不同。
四、Beta 多样性分析
Beta 多样性表示的是微生物群落构成的比较,用来评估微生物群落间的差异。本研究中,我们采用PCoA 分析进行样本的Beta 多样性分析。PCoA 分析,即主坐标分析(principal co-ordinates analysis),是一种非约束性的数据降维分析方法。相比于PCA 分析只能采用欧氏距离算法进行分析计算,PCoA 分析可以通过不同的距离算法来进行分析评估。ANOSIM 分析,即相似性分析(analysis of similarities)是一种非参数检验,用来检验组间(两组或多组)的差异是否显著大于组内差异,其检验值用于PCoA 分析。利用unweighted_unifric算 法(R=0.570,P=0.001)和weighted_unifric 算法(R=0.236,P=0.018)进行PCoA 分析,发现两组样本主成分差异显著(见图3)。
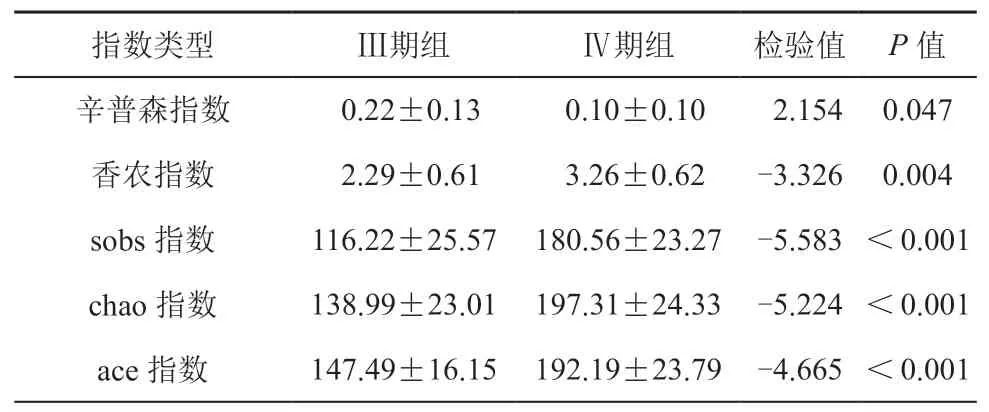
表3 Alpha 多样性指数表
五、组间差异显著性检验
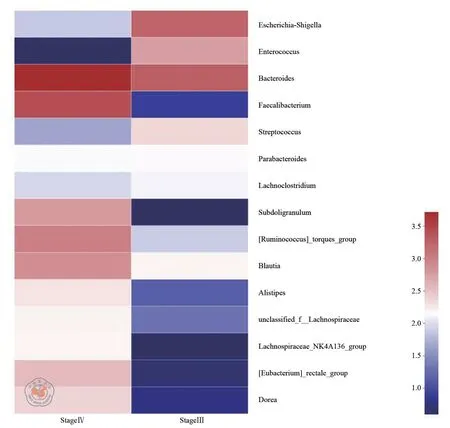
图2 属水平群落Heatmap 图

图3 基于OTU 水平的主坐标分析(3A:unweighted_unifric 算法,R=0.570,P=0.001;3B:weighted_unifric 算法,R=0.236,P=0.018)
组间差异显著性检验根据得到的群落丰富度数据,运用严格的统计学方法来检测不同组(样本)微生物群落中表现出丰富度差异的物种,进行假设性检验,评估观察到的差异的显著性。从门水平看,两组的优势菌门均为厚壁菌门(Firmicutes)、拟杆菌门(Bacteroidetes)和变形菌门(Proteobacteria),Ⅲ期组与Ⅳ期组之间有丰富度显著差异的是疣微菌门(Verrucomicrobia)(见图4)。从属水平看,Ⅲ期组与Ⅳ期组的肠球菌属丰富度差异具有统计学意义(P<0.05),并且Ⅲ期组的肠球菌属丰度更高。此外,两组在Faecalibacterium、Subdoligranulum、Blautia 和Alistipes 等菌属的丰度上亦存在差异(见图5)。为进一步分析肠球菌属丰度的差异对于组间差异的贡献度,我们利用LEfSe 多级物种差异判别分析绘制LDA(线性回归分析)判别柱形图(见图6)。组间显著差异的菌属获得的LDA分值越大,代表物种丰度对差异效果影响越大。结果提示,在组间丰度差异菌属中,肠球菌属对于组间差异贡献最大。
讨 论
人类肠道内定殖着数量众多的微生物群落,其中绝大多数为细菌。近年来,肠道菌群与结直肠疾病及系统性疾病的关联成为了研究热点,关于肠道菌群在结直肠癌发生和发展中的作用机制研究日渐增多[2-8]。多项研究结果表明,进展期结直肠癌与早期结直肠癌的菌群特征有明显差异,大肠杆菌、具核梭杆菌等定殖菌可以促进结直肠癌的进展[5,8,11]。然而,对于进展期结肠癌,不同阶段之间是否存在菌群特征差异尚未见明确报道。因此,我们进行了单中心的临床样本收集,并尝试利用新兴的高通量测序技术探究Ⅲ期与Ⅳ期结肠癌患者的肠球菌属丰度差异,从而揭示不同病理分期的进展期结肠癌患者间菌群特征差异。
近年来,肠球菌属的研究热度逐渐攀升,关于肠球菌属与结直肠癌关联的机制研究及临床队列研究不断增多。然而,目前对于肠球菌属与结直肠癌的关系尚存争议。早期的队列研究发现,与健康人群相比,肠球菌属在结直肠癌患者体内丰度增加,提示肠球菌属可能起到“促瘤”作用[12]。机制研究提示肠球菌属可以产生胞外过氧化物,从而导致肠道上皮细胞DNA 损伤,进而发挥作用[7]。然而,近年来日本学者开展的一项队列研究发现,高丰度的肠球菌属可能与较低的结直肠癌风险相关[6],但亦有其他队列研究提出反对意见[13]。综上,肠球菌属与结直肠癌间确存关联,但究竟是“促瘤”还是“抑瘤”作用尚有争议。此外,鉴于中国人群的临床样本研究结果尚缺乏,我们开展了本项研究,并收集粪便样本来反映肠道菌群特征。

图4 门水平两组肠道菌群差异图

图5 属水平两组肠道菌群差异图
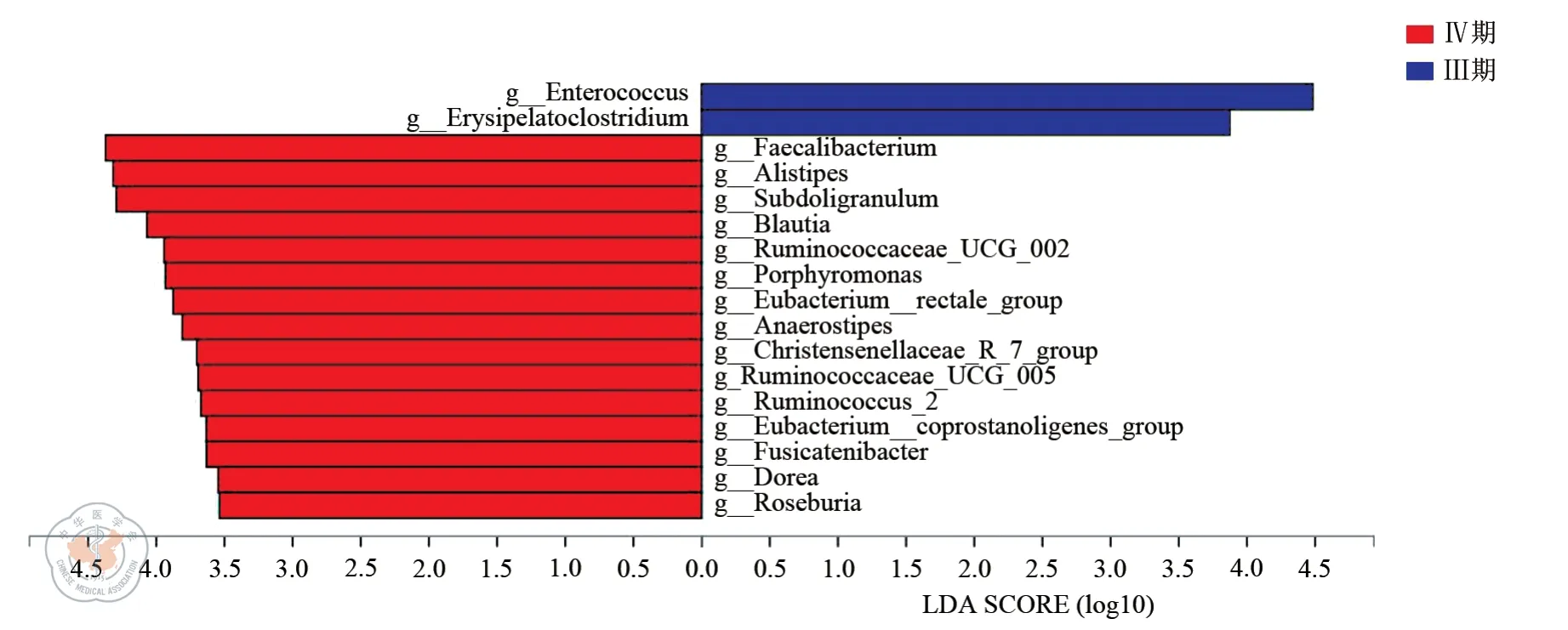
图6 LDA 判别柱形图
粪便样本中包含了大量在粪便形成过程中冲刷下来的肠道黏膜定殖菌。并且,相比于黏膜活检,粪便样本的采集过程属于非侵入性操作,更容易被受检者接受。因而,粪便是研究人体肠道菌群特征的常用样本[14-16]。但侵入性治疗(内镜下治疗及手术治疗)、抗生素治疗、口服泻药或使用灌肠剂等治疗手段都会干扰正常的肠道菌群组成,进而影响粪便样本质量[11,14]。因此我们在入组标准中将近期有上述治疗情况者排除。此外,本研究中倾向值匹配法的使用可以更好地保证基线资料一致,避免基线资料不一致而导致的菌群分析差异结果。
从测序的基本信息来看,测序深度达到了要求,能够支持我们进行后续的生物信息学分析[10]。Alpha 多样性分析提示Ⅲ期组与Ⅳ期组在物种丰富度和多样性上都有明显差异,Ⅳ期组的物种丰富度和多样性发生了明显改变。主坐标分析(PCoA 分析)提示两组菌群的OTU 分布明显不同,进一步的相似性分析(ANOSIM 分析)又证实了组间差异确实大于组内差异,这些结果提示进展期结肠癌的不同阶段,肠道菌群有着明显不同的特征,并且提示本研究的分组有意义。最后,利用组间差异显著性检验,我们发现Ⅲ期组与Ⅳ期组肠球菌属丰度差异显著,并且其丰度差异对组间差异贡献值最高。这可能提示肠球菌属与结肠癌疾病进展存在相关性。
结合既往研究来看,日本学者进行的一项队列研究提示早期CRC 患者与健康人群相比,肠球菌属的丰度降低[6]。并且,该研究发现高丰度的肠球菌属与结直肠癌发生风险降低相关。同时,我们的研究结果又发现Ⅲ期结肠癌患者的肠球菌属丰度较高,肠球菌属的丰度降低对Ⅲ期与Ⅳ期间菌群特征的差异贡献最大,这可能提示肠球菌属丰度的降低与结肠癌疾病进展相关。然而,对于早期结肠癌(Ⅰ期、Ⅱ期)与进展期结肠癌(Ⅲ期、Ⅳ期)之间的菌群差异及肠球菌属发挥的作用尚缺乏研究,无法建立健康人、早期结肠癌到进展期结肠癌与肠球菌属的整体联系,从而论证肠球菌属丰度降低与结肠癌发病及进展相关,这一问题仍有待于更多相关研究的开展。此外,肠球菌属的“抑瘤”角色尚存争议,缺乏机制说明。目前的基础研究发现肠球菌属可通过产生胞外过氧化物,诱导肠道上皮细胞DNA 损伤的方式诱导肿瘤发生[7]。是否肠球菌属可以通过膜表面蛋白结合结肠肿瘤细胞,发挥抑制增殖、促进凋亡的作用,从而发挥“抑瘤”功能?基于本研究成果的后续机制探究可能会获得更加严谨的结论。
综上所述,Ⅲ期结肠癌患者与Ⅳ期结肠癌患者肠道菌群的物种组成和丰富度有显著差异,而肠球菌属丰度改变对差异的贡献最大。这可能提示肠球菌属与结肠癌疾病进展存在相关性。因此,本研究不仅为后续的实验研究提供目标和研究基础,更将有助于为结直肠癌的预防、诊断和治疗提供新思路。
