深圳地区血红蛋白Q-Thailand的表型和基因型分析*
2020-05-30阚丽娟蔡钦泉覃俊龙莫云均张丽军陈亚琼张秀明李育敏
阚丽娟,蔡钦泉,覃俊龙,莫云均,张丽军,陈亚琼,李 瑞,李 悦,张秀明,李育敏
(深圳市罗湖区人民医院检验科,广东深圳 518001)
血红蛋白(Hb)疾病是由于珠蛋白基因缺陷导致的一组疾病,其中引起珠蛋白分子结构改变称为异常Hb疾病,肽链合成不足为珠蛋白生成障碍性贫血[1]。Hb Q-Thailand是由于α珠蛋白肽链α1基因N端第74位密码子GAC→CAC突变,使该位点天门冬氨酸被组氨酸替代所致,属于异常Hb疾病。但由于Hb Q-Thailand通常与-α4.2缺失型珠蛋白生成障碍性贫血连锁遗传,因此,临床对电泳筛查阳性者,应进一步行珠蛋白生成障碍性贫血基因及DNA测序检测以明确基因型,并进行珠蛋白生成障碍性贫血防控指导和优生遗传咨询。莫宗平等[2]对我国广东、广西地区的异常Hb基因突变调查显示Hb Q-Thailand在α链异常的Hb中分布最多。广东省不同地区的研究也表明Hb Q-Thailand的人群携带率较高[3-5]。但是由于不同年龄段及性别的血液学参数不同,且大部分进行珠蛋白生成障碍性贫血筛查的成年女性处于妊娠期,存在生理性贫血的可能,因此,本研究分别对成人和新生儿Hb Q-Thailand的血液学特征进行分析,并按性别分类统计成人的血液学参数指标,为中国人群Hb Q-Thailand的遗传学研究提供系统、详细的临床参考资料。
1 资料与方法
1.1一般资料 选择2016年6月至2019年5月本院门诊和住院进行毛细管电泳检测的病例为研究对象,为连续性资料,共72 397例,分为成年男性、成年女性及新生儿。
1.2仪器与试剂 红细胞参数分析采用血细胞分析仪,购自日本Sysmex公司,型号XN-1000;Hb分析采用全自动毛细管电泳仪,购自法国Sebia公司,型号Capillarys2,试剂均为厂家原装配套试剂。珠蛋白生成障碍性贫血基因检测采用扩增仪,购自中国黑马公司,型号T960;全自动凝胶成像分析仪购自美国Bio-Rad公司,型号ChemiDocTMMP;电泳仪购自中国北京六一仪器厂,型号DYY-6C;恒温杂交仪购自中国深圳亚能生物技术有限公司,型号YN-H16,试剂购自中国深圳亚能生物技术有限公司;DNA测序采用遗传分析仪,购自美国ABI公司,型号3730XL。
1.3方法
1.3.1血液学分析 采集乙二胺四乙酸二钾抗凝的外周静脉全血2 mL,2~8 ℃保存,用于红细胞参数和Hb电泳分析。采用全自动血液分析仪进行红细胞参数分析;采用毛细管电泳法进行Hb组分分析。
1.3.2珠蛋白生成障碍性贫血基因检测 标本采集同血液学分析。采用跨越断裂点PCR法检测3种中国人群常见的缺失型α珠蛋白生成障碍性贫血基因(--SEA、-α3.7和-α4.2);采用PCR结合反向点杂交法检测常见的3种非缺失α珠蛋白生成障碍性贫血基因(αQS、αCS、αWS)和17种β珠蛋白生成障碍性贫血基因点突变类型(-28、-29、CD17、CD41-42、CD43、βE、CD71-72、IVS-Ⅱ-654、-32、-30、CAP、Initiation condon、CD14-15、CD27-28、IVS-Ⅰ-1、IVS-Ⅰ-5、CD31)。
1.3.3珠蛋白基因序列分析 珠蛋白基因测序由中国深圳亚能生物技术有限公司检测。采用Sanger双脱氧链终止法进行珠蛋白基因序列测定,于人类异常Hb和珠蛋白生成障碍性贫血数据库(http://globin.bx.psu.edu)查找突变位点。

2 结 果
2.1基因型分析 经DNA测序,检出成人及新生儿共28例Hb Q-Thailand(GAC→CAC)杂合突变,发生率为0.039%,所有携带者均检出-α4.2杂合缺失型珠蛋白生成障碍性贫血,其中27例为Hb Q-Thailand合并-α4.2杂合缺失,包括成人21例(男性2例,女性19例),新生儿6例;1例为成人Hb Q-Thailand合并-α4.2杂合缺失和βIVS-Ⅱ-81杂合突变(C>T)。见图1、2。
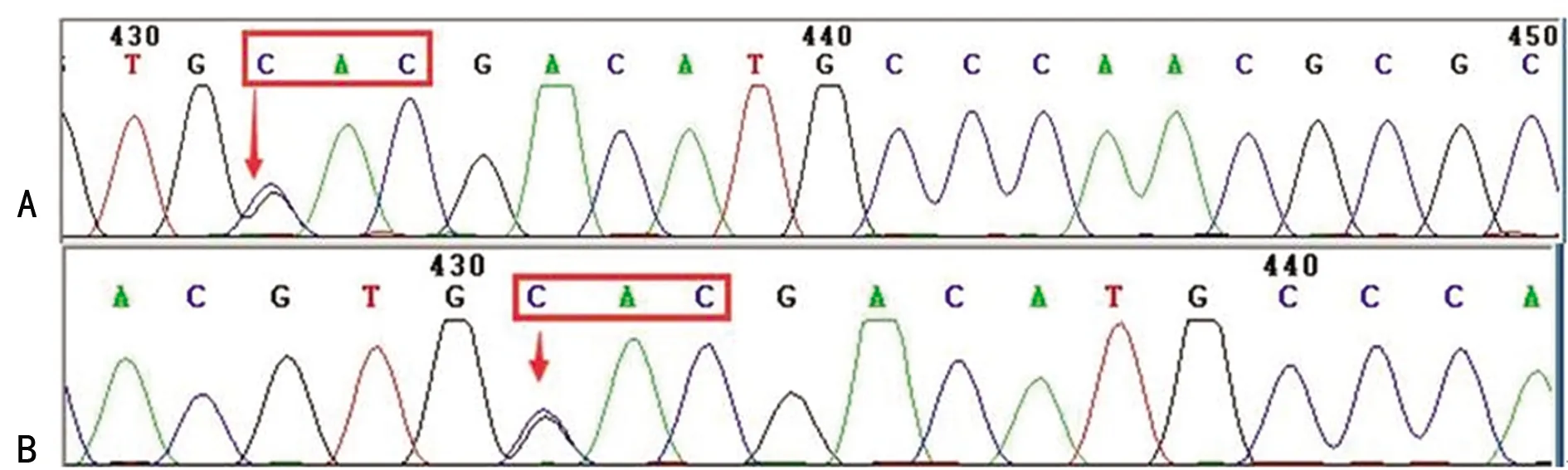
注:A表示成人;B表示新生儿;↓表示突变位点。
图1 成人及新生儿的Hb Q-Thailand测序图
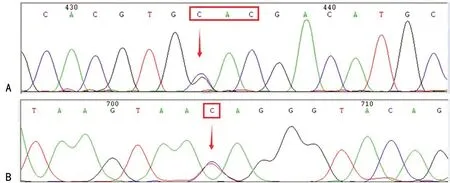
注:A表示Hb Q-Thailand;B表示IVS-Ⅱ-81;↓表示突变位点。
图2 Hb Q-Thailand合并IVS-Ⅱ-81杂合突变测序图
2.2血液学表型分析 成人Hb Q-Thailand的Hb电泳分析均见Hb Q-Thailand和HbA2变异体条带。成人Hb Q-Thailand合并-α4.2杂合缺失,以及合并βIVS-Ⅱ-81杂合突变的红细胞参数分析均表现为Hb水平正常,平均红细胞体积(MCV)和平均红细胞血红蛋白量(MCH)轻度降低。1例成人男性Hb Q-Thailand合并IVS-Ⅱ-81杂合突变,Hb 120 g/L,MCV 68.0 fL,MCH 23.7 pg,HbA2+突异体2.4%,Hb Q-Thailand 28.3%。6例新生儿Hb Q-Thailand杂合子的Hb电泳结果均见Hb Q-Thailand和Hb Bart′s带。6例新生儿平均HbA(13.7±4.1)%,HbF(64.9±4.1)%,Hb Q-Thailand(21.0±2.7)%,Hb Bart′s(0.4±0.1)%。见表1、2。
表1 成人Hb Q-Thailand的血液学表型
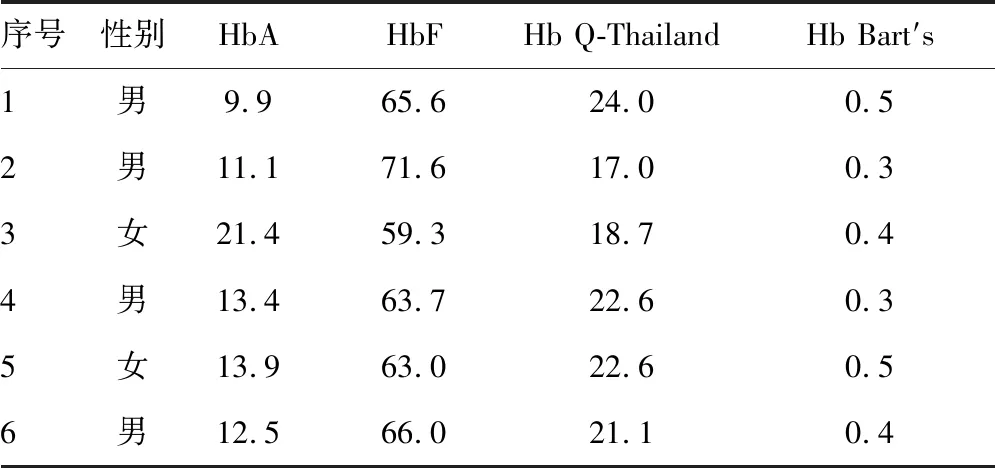
表2 6例新生儿Hb Q-Thailand杂合子的毛细管电泳分析(%)
3 讨 论
Hb Q-Thailand是1958年由VELLA等[6]在东南亚1个华侨家庭中发现。曾溢滔等[7]于1983年在我国江西萍乡首先发现Hb Q-Thailand携带者。我国广东、广西地区异常Hb以HbE、Hb Q-Tailand、Hb New York为主[2]。广西南宁地区调查Hb Q-Thailand的发生率为0.130%[8],柳州为0.078%[9],而李友琼等[10]的研究报道广西地区为0.060%,均较高;广东省调查显示韶关为0.164%[3],梅州为0.085%[4],而广州市针对育龄期夫妇的调查显示为0.171%[5]。本研究纳入深圳地区72 397例标本进行大规模的群体异常Hb疾病筛查,显示Hb Q-Thailand的发生率为0.039%,低于以上所有地区,这可能跟深圳作为移民城市,受人口迁徙影响和基因构成比例复杂等因素有关。
Hb Q-Thailand通常与-α4.2缺失型珠蛋白生成障碍性贫血连锁遗传有关,但是由于其突变位点天门冬氨酸是位于分子表面非功能位置的氨基酸,被组氨酸代替不影响Hb的功能,因此,其合并-α4.2缺失杂合子常无明显贫血症状,血细胞分析表现为Hb水平正常,MCV和MCH轻度降低,少部分携带者红细胞参数可正常[8-10]。Hb Q-Thailand合并α珠蛋白生成障碍性贫血时,Hb Q-Thailand的量与α基因的缺失个数呈正相关,α基因缺失数量越多,Hb Q-Thailand的水平越高[2,8]。Hb Q-Thailand合并轻型α珠蛋白生成障碍性贫血时(如同时合并-α4.2缺失和-α3.7缺失,或合并-α4.2缺失纯合子)的血液学表型同轻型α珠蛋白生成障碍性贫血[8-10]。但如Hb Q-Thailand合并中间型α珠蛋白生成障碍性贫血,则会引起Hb Q-H病。Hb Q-H病的主要基因型为α0/α+,即3个α基因失活,而剩下的1个α基因发生突变形成Hb Q-Thailand。因此,成人Hb Q-H病可无HbA和HbA2合成,而合成Hb Q-Thailand(αQ2β2)和Hb QA2(αQ2δ2)及过剩的β链形成的Hb H,电泳表现Hb以Hb Q-Thailand为主,然后为Hb H、Hb Bart′s和少量Hb QA2[11-12];产前诊断脐带血标本表现为Hb Q-Thailand和Hb Bart′s及少量的Hb portland[13]。由于Hb Q-Thailand稳定且有携氧能力,大部分报道显示Hb Q-H病与Hb H病的临床症状相似[9,11],但也有报道表明Hb Q-H病严重者可导致胎儿水肿[13],可能与Hb Q-Thailand合并Hb H病加重缺氧有关,这提示临床如夫妻一方为Hb Q-Thailand合并-α4.2缺失,另一方为α0基因(如--SEA和-THAI)携带者,母亲在妊娠期有发生胎儿水肿的可能,必要时仍需进行产前诊断。Hb Q-Thailand合并-α4.2缺失合并轻型β珠蛋白生成障碍性贫血的血液学表型同轻型β珠蛋白生成障碍性贫血,但Hb Q-Thailand水平低于Hb Q-Thailand合并-α4.2缺失杂合子,这可能是由于β链合成降低导致Hb Q-Thailand减少[10-14]。Hb Q-H病合并轻型β珠蛋白生成障碍性贫血,血液学表型与Hb Q-H病相似,表现为轻至中度小细胞低色素性贫血,Hb Q-Thailand明显升高,但前者HbA2升高[8,15]; Hb Q-H病合并中间型β珠蛋白生成障碍性贫血也与Hb Q-H病相似,表现为轻至中度小细胞低色素性贫血,这可能是由于α链和β 链均合成降低,使α链/β链失衡减少[11]。偶有Hb Q-Thailand不与-α4.2缺失型珠蛋白生成障碍性贫血连锁遗传的报道,1例为Hb Q-Thailand合并--SEA缺失和βCD41-42杂合子,表现为轻型珠蛋白生成障碍性贫血,Hb Q-Thailand水平较Hb Q-Thailand合并-α4.2缺失杂合子低,HbA2升高,这与β链合成减少有关[8];另外2例为单纯Hb Q-Thailand杂合子,Hb Q-Thailand水平为(18.9±1.8)%[2]。
本研究检出的Hb Q-Thailand均为杂合子,并与-α4.2缺失型珠蛋白生成障碍性贫血连锁遗传,其中成人携带者以女性居多,这是由于进行产前Hb电泳筛查的妊娠期女性所占比例较高。鉴于妊娠期存在生理性贫血的可能,本研究首先按性别分别统计成人的各项血液学参数指标,观察到成年男性和女性Hb Q-Thailand合并-α4.2杂合缺失携带者均无明显贫血症状,Hb水平在正常参考区间内,仅表现为MCV和MCH轻度降低,这表明MCV和MCH在珠蛋白生成障碍性贫血筛查中的重要性;其中2例携带者红细胞参数正常,与已有报道相符合[10],这也提示临床在珠蛋白生成障碍性贫血筛查时需要结合血常规和Hb电泳结果联合分析,避免漏诊。本研究还发现1例成人Hb Q-Thailand合并-α4.2杂合缺失和βIVS-Ⅱ-81杂合突变,其红细胞参数特征同Hb Q-Thailand合并-α4.2杂合缺失。HbVar数据库上记录β珠蛋白基因IVS-Ⅱ-81(C>T)突变(HBB:c.315+81C>T)发生于印度人群,可能为中性多态性位点。笔者也曾报道1例Hb Queens合并IVS-Ⅱ-81杂合突变,其血液学表型正常,这表明IVS-Ⅱ-81杂合突变不影响β珠蛋白基因的功能[16]。新生儿珠蛋白生成障碍性贫血筛查也是珠蛋白生成障碍性贫血防控的重要环节之一,研究表明脐带血Hb电泳分析在新生儿珠蛋白生成障碍性贫血筛查中较传统的红细胞脆性试验及血细胞分析更具筛查价值[17],因此,新生儿通常采用脐带血Hb电泳进行珠蛋白生成障碍性贫血筛查。本研究分析新生儿Hb Q-Thailand的Hb电泳特征,6例新生儿Hb Q-Thailand合并-α4.2杂合缺失的脐带血均见Hb Q-Thailand和Hb Bart′s带,平均水平分别为(21.0±2.7)%和(0.4±0.1)%。王继成等[13]报道Hb Q-H病的产前诊断脐带血标本Hb Q-Thailand和Hb Bart′s的水平分别为(46.6±6.1)%和(32.7±6.7)%,其原因同成人外周血Hb合成,即3个α基因失活,只剩下1个带Hb Q-Thailand的α1基因。
4 结 论
综上所述,本研究按年龄段及性别分类统计成人及新生儿Hb Q-Thailand的血液学参数指标,为珠蛋白生成障碍性贫血筛查提供了系统的临床参考资料,并结合研究结果和文献综合分析了Hb Q-Thailand合并不同珠蛋白生成障碍性贫血的血液学特征,这对于Hb Q-Thailand的遗传学研究具有重要的参考价值。
