从二苯胺到咔唑取代:芘类化合物发光性质的理论研究
2020-05-22宋晓娟巩光帅王传增赵保忠张莉莉
宋晓娟 褚 赛 巩光帅 王传增 赵保忠 张莉莉 张 天
(山东理工大学 化学化工学院,山东 淄博 255049)
0 引言
芘是由四个苯环稠合而成的多环芳烃,最初被用于合成染料[1].现如今,芘类化合物在有机电子、化学传感和生物成像等领域都得到了广泛应用[2,3].为了进一步开发高效的有机光电子器件,调控芘类化合物的发光性质引起了研究工作者的兴趣.Yamato等人报道了一种给体-受体(D-A)型芘类荧光分子,D为二苯胺(DPA),A为叔丁基芘(PY),即2DPA-PY(如图1所示).实验结果表明,在二氯甲烷溶液中2DPA-PY的荧光量子产率(Φf)为82.0%,且在DPA的苯环对位引入给电子基团(Me、tBu、OMe),可以调控衍生物的发光颜色从蓝色到黄色[4].然而,溶液下衍生物的Φf却随着取代基给电子能力的增强而减小.最近,我们又在相同位置添加了吸电子基团(F、CF3、CN)并开展了第一性原理研究,发现溶液下衍生物的Φf随着取代基吸电子能力的增强而增强,还预测出CN衍生物的Φf最高可以达到89.6%[5].由此可见,通过降低D-A型分子中给体的给电子能力,可以有效提高溶液下这类化合物的Φf.本文我们不再沿用DPA的给体,而是将其直接替换为给电子能力更弱的咔唑(CBZ)[6,7],即设计出2CBZ-PY(如图1所示),以期得到更高的发光效率.通过对比研究2DPA-PY和2CBZ-PY 的电子结构和激发态性质,从而得出这类D-A型芘类化合物分子结构和发光性能之间的关系.
1 理论部分
我们分别采用密度泛函理论(DFT)[8,9]和含时密度泛函理论(TD-DFT)[10]对分子的基态(S0)和第一单重激发态(S1)进行电子结构计算.使用PBE0[11]泛函和6-31G(d)基组进行结构优化,该计算水平已成功应用于描述有机荧光分子的单重激发态[12,13].然而,由于PBE0这类传统泛函往往会低估分子内电子转移(ICT)态的激发能[14],所以我们在PBE0优化好的几何结构基础上,再用MPW1B95[15]/6-31G(d)水平计算分子的跃迁性质.二氯甲烷的溶剂化效应通过极化连续介质模型(PCM)来模拟.上述计算是在Gaussian 16软件[16]中完成的.基于上述计算得到的电子结构和振动信息,我们再用热振动关联函数方法计算振动分辨的吸收、发射光谱和发光量子效率.

吸收光谱和发射光谱的解析表达式[17]可以分别写作
(1)
(2)
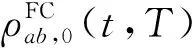
(3)
基于费米黄金规则得到的knr表达式[17,18]可以写作
(4)
其中ρic,kl(t,T)是内转换过程的热振动关联函数,Zi是配分函数.Rkl表示非绝热电子耦合矩阵元,可以根据林圣贤等人的一阶微扰理论[19]得到.为确保关联函数的收敛,两分子knr的计算均采用了洛伦兹展宽(FWHM=10.62 cm-1).kr和knr是在MOMAP程序[20]中计算得到的.
2 结果与讨论
2.1 几何结构

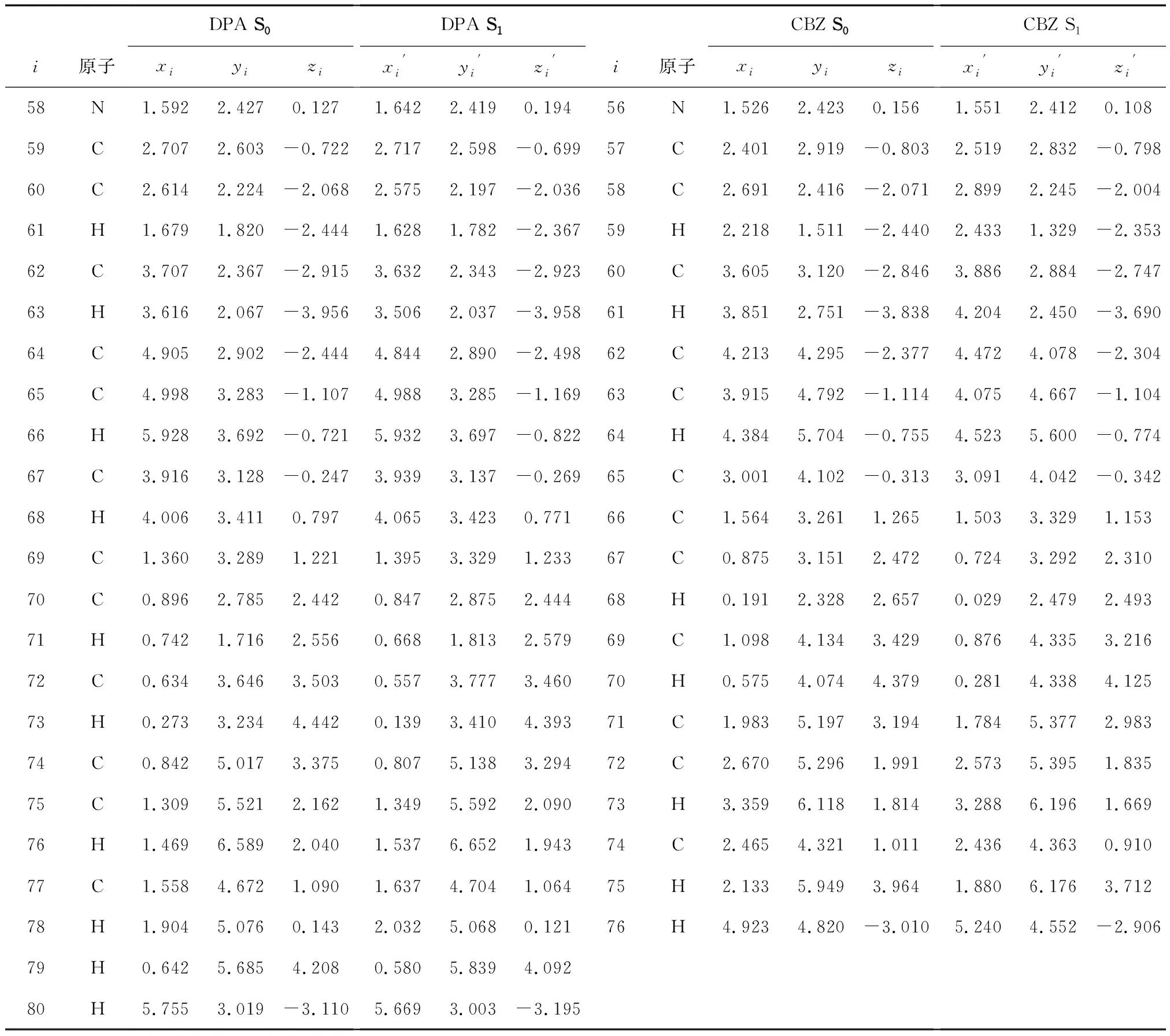
表1 取代基部分的原子坐标
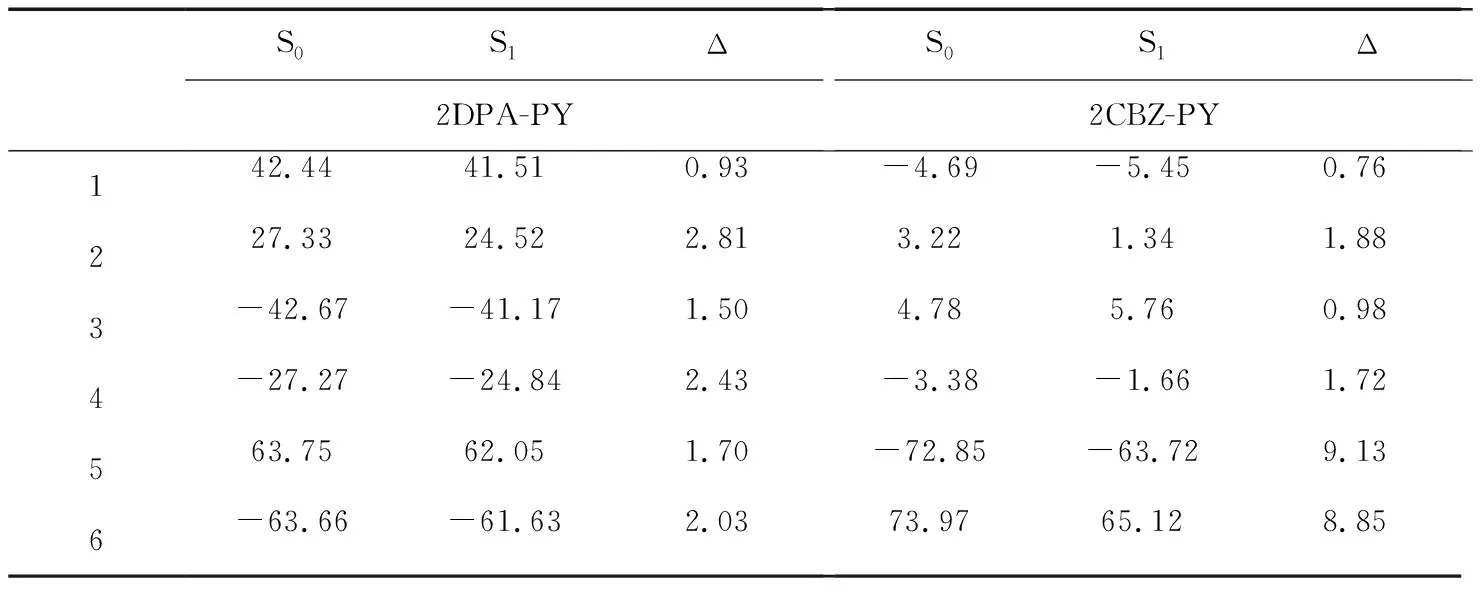
表2 重要二面角(以度为单位)
2.2 跃迁性质
在表3中我们列出了2DPA-PY和2CBZ-PY的跃迁性质.计算所得的2DPA-PY的垂直激发能ΔEvert与已有实验值(exp.)吻合地很好,计算值与实验测得的吸收和发射峰的偏差仅分别为0.08 eV和0.01 eV,斯托克斯位移(即吸收和发射峰能量的差值)为0.27 eV.而与2DPA-PY相比,理论预测的2CBZ-PY的吸收和发射峰均发生了蓝移,且斯托克斯位移更大,为0.32 eV.从表3可知,它们的跃迁成分主要是HOMO→LUMO,表现出明显的ICT特征(如图3所示).2CBZ-PY的HOMO-LUMO能差比2DPA-PY大,因而发光波长会发生蓝移.2CBZ-PY的电子跃迁偶极矩μ0小于2DPA-PY,表明其HOMO和LUMO轨道的重叠小于2DPA-PY,这是由CBZ较大的位阻所引起的[22].而两者的振子强度f却非常接近,这是因为f不仅与μ02成正比,还与两个态之间的能量差成正相关.尽管2CBZ-PY的μ0小于2DPA-PY,ΔEvert却大于2DPA-PY.
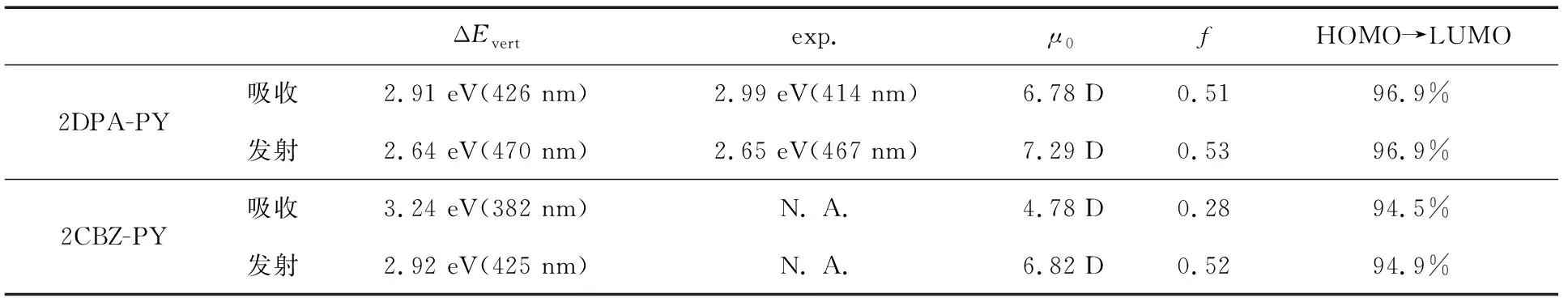
表3 垂直激发能(ΔEvert)、实验值(exp.)、电子跃迁偶极矩(μ0)、振子强度(f)和跃迁成分(HOMO→LUMO)
2.3 重整能
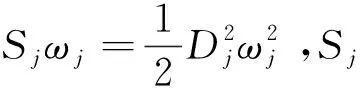
2.4 发光效率
计算得到的室温下振动分辨的吸收和发射光谱如图5所示.谱线的展宽源于分子的振动结构和温度效应,计算中并没有额外引入展宽因子.2CBZ-PY的吸收和发射光谱相对于2DPA-PY均发生了明显的蓝移,这与我们先前计算的垂直跃迁性质是一致的.在表4中我们列出了计算所得的kr、knr和Φf.2DPA-PY的Φf计算值与已有实验值相差很小,这进一步说明了计算方法的合理性.理论预测的2CBZ-PY的kr比2DPA-PY大,knr比2DPA-PY小.由爱因斯坦自发辐射关系[24,25]可知,kr与f和ΔEvert的平方成正比.尽管2CBZ-PY的f与2DPA-PY非常接近(见表3),但其ΔEvert大于2DPA-PY,故2CBZ-PY的kr大.为了更深入地理解它们的无辐射衰减过程,我们画出了两者的无辐射谱线(如图6所示).当能隙ΔE等于绝热激发能ΔEad时,对应的纵坐标即为log(knr).根据能隙定律[17,19],当ΔE足够大的时候,log(knr)几乎随着ΔE的增加而线性减小.即ΔEad越大,knr越小.2DPA-PY和2CBZ-PY的ΔEad分别是2.78 eV和3.07 eV,故2CBZ-PY的knr小.因此,增大的kr和减小的knr使得2CBZ-PY的Φf比2DPA-PY大,其计算值为93.8%,比我们之前通过在DPA对位引入吸电子基团所得的衍生物还要高.

表4 室温下的kr、knr和Φf(括号为实验值)
3 结论
本文采用热振动关联函数方法和极化连续介质模型,研究了溶液下两种D-A型芘类化合物2DPA-PY和2CBZ-PY的发光性质.研究表明,将给体从二苯胺替换为给电子能力更弱的咔唑后,2CBZ-PY比2DPA-PY的结构变化程度和斯托克斯位移更大.通过对重整能的分析,我们找出了它们在激发态弛豫过程中的能量耗散通道.2CBZ-PY的发射波长相对于2DPA-PY蓝移,根据爱因斯坦自发辐射关系可得其辐射速率增大.2CBZ-PY的绝热激发能比2DPA-PY大,由能隙定律可知其无辐射速率减小.加快的辐射过程和减慢的无辐射过程使得2CBZ-PY的发光效率比2DPA-PY更高,其理论预测值为93.8%.
