抑制低密度脂蛋白受体相关蛋白2表达对Alzheimer’s病细胞模型丝裂原活化蛋白激酶信号通路影响的机制研究
2020-05-18王丽玲李焰生王鹏飞
王丽玲,李焰生,王鹏飞
Alzheimer’s病(AD)是老年人最常见的神经系统退行性疾病,临床表现为记忆和认知功能不断恶化,日常生活能力进行性减退,并可伴有各种神经精神症状[1-2]。国际AD协会针对全球AD对经济影响的评估报告称,2018年全球约有5 000万人患有痴呆,到2050年这一数字将增至1.52亿,是现在的三倍之多。据估计,2018年全球社会痴呆相关成本为1万亿美元,到2030年这一数字将增至2万亿美元[3]。AD的神经病理改变大体表现为以海马和皮质为主的全脑萎缩,脑沟回增宽及脑室增大,镜下主要表现为神经元外由β淀粉样蛋白(Aβ)沉积形成的老年斑以及神经元内由tau蛋白过度磷酸化形成的神经原纤维缠结(NFTs)[4]。迄今为止,已有许多AD的假说,主要包括淀粉样蛋白假说、tau蛋白过度磷酸化假说、神经元凋亡假说、自由基损伤及免疫炎症假说、神经递质代谢障碍假说等。但任一假说都不足以全面阐述AD的发病机制。越来越多的证据显示,遗传、环境和衰老因素共同参与了AD的发生发展[5]。
低密度脂蛋白受体(LDLR)是一种进化上古老而又高度保守的基因家族,属于Ⅰ型跨膜受体,在结构上具有高度相似性。所有成员胞外段都有配体结合域、表皮生长因子样富含半胱氨酸重复序列和包含β螺旋的YWTD基序,同时也都含有一个单次跨膜区及50~200个氨基酸不等且有NPXY基序的胞内段,胞内段可能参与受体内吞和信号转导的调控。低密度脂蛋白受体相关蛋白(LRP)1是LDLR家族第一个被发现的成员,广泛表达于全身各组织器官,尤其是脑组织,在神经元、小胶质细胞及星形胶质细胞中均存在表达。病理研究证实,脑组织LRP1的表达在AD患者中显著下降,LRP1在脉络丛上皮细胞中可以直接与Aβ结合,从而促进其在血-脑屏障中的清除[6]。研究发现,在原代神经元细胞中干扰LRP1的表达可导致神经元细胞的凋亡增加[7]。在小鼠前脑神经元中特异性地敲除LRP1可引起脂质代谢异常,并导致进行性年龄依赖的树突棘退化、突触丢失、神经炎症、小鼠记忆功能下降等全面神经退行性病变[8]。而同样属于LDLR家族的LRP2在结构上与LRP1非常相似,是一种广泛表达与全身的多配体受体。既往研究认为,LRP2主要表达于室管膜细胞、毛细血管和脉络丛上皮细胞[9],在脑毛细血管和脉络丛上皮参与ApoJ和Aβ复合物的摄取、转运及降解[9-11],并且LRP2在脉络膜丛中的表达及其功能随年龄增长、AD发生而降低[12-14]。此外,LRP2在脉络丛有助于类胰岛素生长因子1(IGF-1)、瘦素的转运入脑组织,从而发挥脑保护作用[12-13,15]。上皮细胞选择性敲除LRP2小鼠呈现明显的焦虑行为,学习能力受损和记忆障碍,与AD的症状十分相似[16]。本研究小组既往研究显示,rs3755166多态性可能是中国大陆汉族人群AD发病的危险因素,rs3755166位点由G突变为A后,LRP2基因转录活性明显下降,可能是其导致AD易感的机制[17]。这些都说明LRP2在AD发病及进程中具有重要作用和研究价值。
越来越多的证据表明,LRP2不仅表达于室管膜细胞、毛细血管和脉络丛上皮等细胞中,亦广泛表达于皮质及海马等神经元中[18]。然而,目前对于LRP2在神经元细胞中的功能知之甚少。Aβ是老年斑的主要成分,大量的研究证明过量的Aβ可以通过激活caspase-3引起神经元细胞的凋亡[19-20]。丝裂原活化蛋白激酶(MAPKs)信号通路与细胞凋亡密切相关,该通路参与AD的发病发展[21]。那么在Aβ处理的AD细胞模型中的LRP2表达是否存在异常?由于LRP2与LRP1结构相似,亦是多配体受体,它是否会如同LRP1一样在神经元细胞中具有抗凋亡作用发挥对细胞的保护性作用?在神经元细胞中调控LRP2的表达对神经元细胞及其对Aβ诱导的细胞凋亡是否具有调控作用呢?LRP2与MAPK信号通路直接存在何种关系呢?基于以上这些问题本研究展开了深入的探究。
1 材料与方法
1.1 材料
1.1.1 实验材料及分组 人神经母细胞瘤(SH-SY5Y)细胞购于American type culture collection,分为空白对照组、阴性对照(SC)组、LRP2 siRNA-1(siLRP1)组、LRP2 siRNA-2(siLRP2)组、siLRP2+Aβ组和SC+Aβ组。
1.1.2 常用试剂 JNK抑制剂SP600125(美国Cell signaling Technology公司);PrimeScript 1st Strand cDNA Synthesis Kit(大连宝生物工程有限公司),anti-Lrp2(美国Abcam公司),anti-GAPDH(AF0006,碧云天生物技术),anti-cleaved Caspase-3(Merck Millipore公司),anti-Caspase-3(8G10)(美国Cell signaling Technology公司),anti-p-ERK1/2(美国Cell signaling Technology公司),anti-ERK1/2(美国Cell signaling Technology公司),anti-p-JNK(美国Cell signaling Technology公司),anti-JNK(美国Cell signaling Technology公司),anti-p-p38 MAPK(美国Cell signaling Technology公司),anti-p38 MAPK(美国Cell signaling Technology公司),羊抗鼠IgG(HRP标记)抗体(美国Sigma公司),羊抗兔IgG(HRP标记)抗体(美国Sigma公司)。
1.2 方法
1.2.1 细胞培养 人神经母细胞瘤细胞(SH-SY5Y细胞)培养于含10%胎牛血清、1%的青-链霉素混合物的高糖DMEM细胞培养液内,放置于37 ℃、5%的二氧化碳培养箱中。六孔板种植SH-SY5Y细胞,培养24 h,待融合度为70%时进行转染,转染48 h后更换培养液。
1.2.2 细胞活力的测定及Aβ1-42处理浓度的选择 不同浓度(0 μmol、5 μmol、10 μmol、20 μmol、30 μmol)Aβ1-42(购于Sigma公司,ddH2O溶解后37 ℃培养箱放置3 d,形成聚集态)处理SH-SH5Y细胞24 h后,选择细胞活力下降50%的Aβ1-42浓度作为AD细胞模型处理浓度。采用噻唑蓝比色法(MTT)测定细胞活力。将细胞种植在96孔板中,密度为2 000 cells/每孔,边缘孔用PBS充填减少蒸发。在5%的CO2孵育箱中37 ℃培养24 h。第2 d细胞贴壁后去培养液,每孔加入90 μl培养液和10 μl 5 mg/L的MTT,37 ℃孵育4 h。小心吸去MTT,加入200 μl DMSO溶解结晶。室温摇晃避光孵育15 min后用多功能酶标仪测定,设定波长为570 nm,参考波长为630 nm。在检测LRP2干扰后对细胞活力影响时,以(转染组-空白对照组)/(SC组-空白对照组)比值作为相对细胞活力。独立实验重复3次,每次复孔在3个以上。
1.2.3 LRP2 siRNA转染方法 LRP2 siRNA由上海吉玛制药技术有限公司设计合成,序列如下:siRNA-1:5′-GUG GAG AAA UGA CAG UUA UTT-3′,5′-AUA ACU GUC AUU UCU CCA CTT-3′;siRNA-2:5′-GGA CUU GUG UUG AUA UUG ATT-3′,5′-UCA AUA UCA ACA CAA GUC CTT-3′;Scrambled siRNA:5′-UUC UUC GAA CGU GUC ACU TT-3′,5′-ACG UGA CAC GUU CGG AGA ATT-3′。将SH-SY5Y细胞用胰酶消化后按1∶3接种至孔板培养,待细胞长至约70%融合度时进行转染,转染具体操作参照脂质体Lipofectamine 2000说明书进行。
1.2.4 LRP2 mRNA水平的检测 于转染24 h及48 h采用RT-PCR检测各组LRP2 mRNA水平。Trizol(Invitrogen公司)提取总RNA,采用逆转录引物用试剂盒(大连宝生物工程有限公司)进行逆转录反应。Homo LRP2及GAPDH基因序列由NCBI neucleotide数据库获得,PCR引物通过Primer 5.0软件设计,并经Primer Blast比对序列正确且特异,由上海英俊生物技术公司合成。引物序列:(1)LRP2引物序列(5′-3′):F:TAG GGC TTA TGT TCT GGA CT,R:TGG GCA CTG ACT TAT TGT AT;(2)GAPDH引物序列(5′-3′):F:CGG AGT CAA CGG ATT TGG TCG TAT,R:AGC CTT CTC CAT GGT GGT GAA GAC。PCR反应体系参照PCR反应试剂盒(Takara公司)说明书,LRP2 PCR反应条件:95 ℃预变性5 min→95 ℃变性30 s→ 55 ℃退火30 s→72 ℃延伸30 s,扩增35个循环后于72 ℃再延伸10 min。GAPDH PCR反应条件:95 ℃预变性5 min→95 ℃变性30 s→ 60 ℃退火30 s→72 ℃延伸30 s,扩增28个循环后于72 ℃再延伸10 min。PCR产物琼脂糖凝胶电泳分析,Image J软件分析条带灰度值。
1.2.5 细胞凋亡的检测 六孔板种植SH-SY5Y细胞,培养24 h待融合度为70%时进行转染,转染48 h后更换培养液。每孔加入20 μmol Aβ1-42处理24 h,异硫氰酸酯(FITC)Annexin V Apoptosis Detection试剂盒(美国BD公司)染色,流式细胞仪(美国BD公司)检测细胞凋亡率。
1.2.6 LRP2、Caspase-3、Anti-Caspase 3,Cleaned Caspase 3蛋白表达水平的检测 于LRP2 siRNA转染48 h后采用Western blotting法检测LRP2蛋白表达水平,LRP2 siRNA转染48 h,并予Aβ1-42处理24 h后,采用Western blotting法检测Caspase-3、Anti-Caspase 3,Cleaned Caspase 3蛋白表达水平。RIPA蛋白裂解液提取蛋白,BCA试剂盒测定蛋白浓度。取40 μg蛋白,加至10% SDS-聚丙烯酸胺凝胶电泳,转膜至PVDF膜。5% BSA封闭,加入一抗anti-Lrp2(1∶1 000)、anti-GAPDH(1∶3 000)、anti-Caspase 3,Cleaned Caspase 3(1∶1 000)、anti-Caspase-3(8G10)(1∶1 000),4 ℃孵育过夜,TBST洗膜,加入相应的二抗。室温封闭2 h后,TBST洗膜,在膜上加入新鲜配置的显影液(Millipore公司),然后采用ChemiDocTMMP imaging system(Bio-Rad公司)进行显影和分析。
1.2.7 MAPKs信号通路(ERK、p38以及JNK磷酸化)表达水平的检测 siLRP2组、SC组、siLRP2+Aβ组和SC+Aβ组细胞采用JNK抑制剂SP600125(200 ng/ml)预处理1 h,随后与Aβ1-42共处理细胞24 h。Western blotting法检测各组(p-ERK1/2)/ERK、p-p38/p38和p-JNK/JNK,稀释比例为1∶1 000。
2 结 果
2.1 干扰LRP2后对AD细胞生存活力的影响 以5 μmol、10 μmol、20 μmol、30 μmol Aβ1-42处理组细胞活力分别为80%、63%、52%、42%。与空白对照组比较,10 μmol、20 μmol、30 μmol浓度Aβ1-42处理的SH-SH5Y细胞活力显著下降(均P<0.05)。Aβ1-4220 μmol时SH-SY5Y细胞活力下降明显(52%),因此选择20 μmol作为AD细胞模型处理浓度。LPR2 siRNA转染48 h后给予20 μmol Aβ处理24 h,siLRP2+Aβ组细胞活力(40.7±1.9%)显著低于SC+Aβ组(54.5±7.5%)(P<0.05)。
2.2 Aβ对SH-SY5Y细胞LRP2蛋白表达水平的影响 见图1。Aβ处理前,SH-SY5Y细胞LRP2/GAPDH蛋白相对表达水平为100%,Aβ1-42(20 μmol)处理后LRP2/GAPDH蛋白相对表达水平为(77.3±15.3)%。与处理前比较,Aβ1-42处理后LRP2蛋白表达水平显著降低(P<0.05)。

图1 LRP2蛋白表达电泳图
2.3 各组LRP2 mRNA及蛋白表达的比较 见表1、图2。与空白对照组比较,siRNA-1组、siRNA-2组LRP2 mRNA及蛋白表达水平均显著降低(P<0.05~0.01),SC组差异无统计学意义(P>0.05)。
2.4 SC+Aβ组及siLRP2+Aβ组细胞凋亡率的比较 见图3。SC+Aβ组细胞凋亡率[(32.4±3.0)%]显著低于siLRP2+Aβ组[(51.8±4.2)%](P<0.01)。
2.5 抑制LRP2表达对MAPKs信号通路的影响 见表2、图4。SC+Aβ组及siLRP2+Aβ组(p-ERK1/2)/ERK与SC组及siLRP2组比较,差异无统计学意义(均P>0.05)。与SC+Aβ组相比,siLRP2+Aβ组(p-ERK1/2)/ERK差异也无统计学意义(P>0.05)。SC+Aβ组及siLRP2+Aβ组p-p38/p38显著高于SC组及siLRP2组(均P<0.01)。与SC+Aβ组相比,siLRP2+Aβ组p-p38/p38差异无统计学意义(P>0.05)。SC+Aβ组及siLRP2+Aβ组p-JNK/JNK显著高于SC组及siLRP2组(均P<0.01)。与SC+Aβ组比较,siLRP2+Aβ组p-JNK/JNK显著增高(P<0.01)。

表1 各组LRP2 mRNA及蛋白相对表达(%)组别LRP2 mRNALRP2蛋白SC组98.5±16.196.8±1.6siRNA-1组48.8±26.9▲58.7±1.4▲siRNA-2组65.8±26.1△66.7±4.3▲空白对照组100100 注:与空白对照组比较△P<0.05,▲P<0.01

图2 各组SH-SY5Y细胞LRP2表达情况 A:各组LRP2 mRNA电泳图;B:各组LRP2蛋白电泳图

图3 SC+Aβ组及siLRP2+Aβ组细胞凋亡率 左下象限表示FITC-/碘化丙啶(PI)-,代表正常细胞;右下象限表示FITC+/PI-,代表早期凋亡细胞;左上象限表示FITC-/PI+,代表死亡细胞;右上象限表示FITC+/PI+,代表晚期凋亡细胞
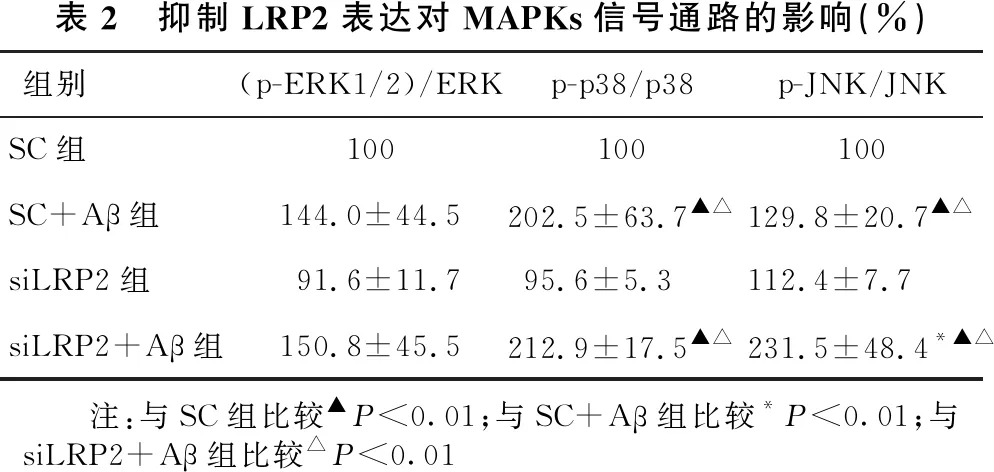
表2 抑制LRP2表达对MAPKs信号通路的影响(%)组别(p-ERK1/2)/ERKp-p38/p38p-JNK/JNKSC组100100100SC+Aβ组144.0±44.5202.5±63.7▲△129.8±20.7▲△siLRP2组91.6±11.795.6±5.3112.4±7.7siLRP2+Aβ组150.8±45.5212.9±17.5▲△231.5±48.4*▲△ 注:与SC组比较▲P<0.01;与SC+Aβ组比较*P<0.01;与siLRP2+Aβ组比较△P<0.01
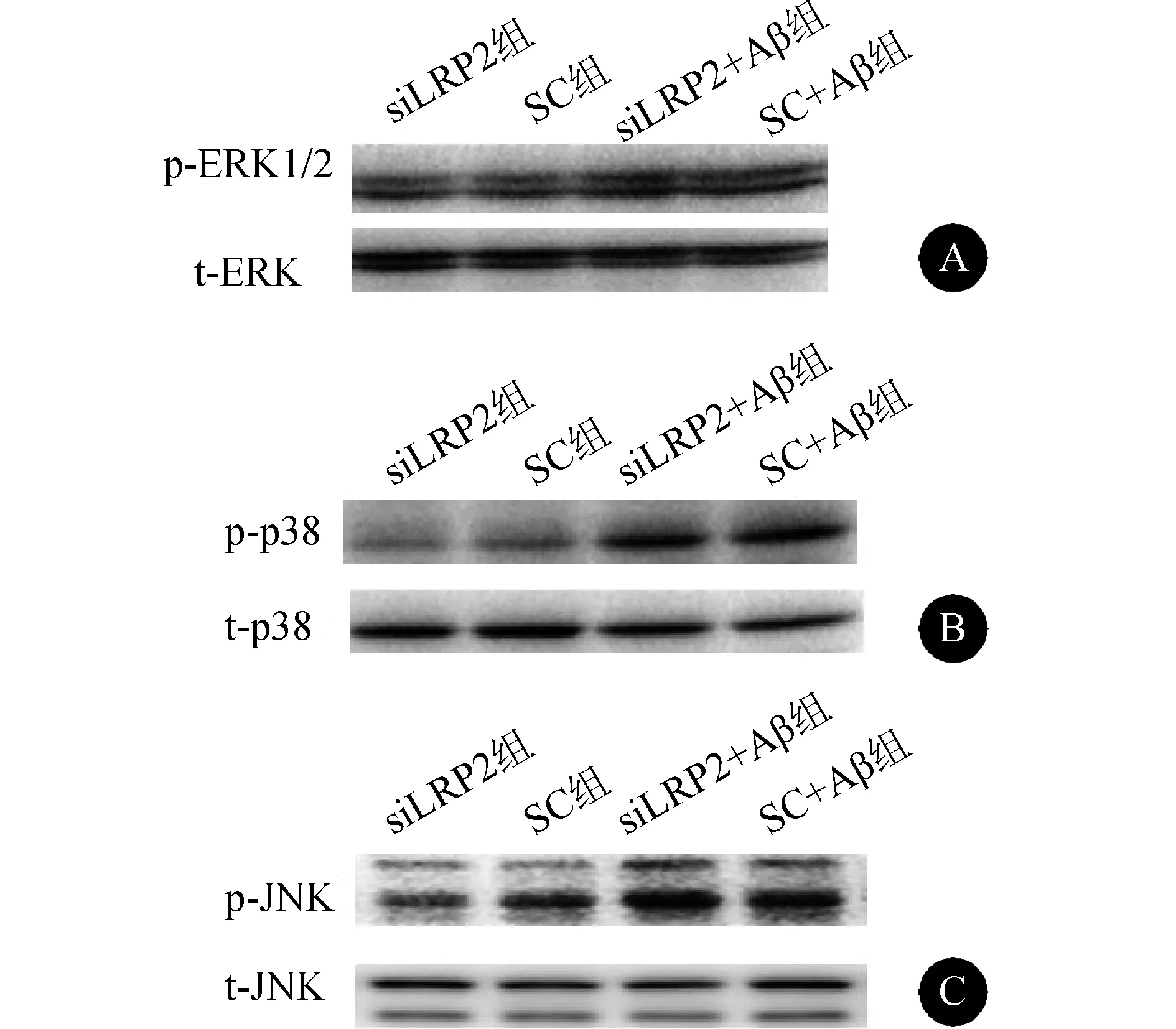
图4 抑制LRP2表达对MAPK信号通路的影响 A:(p-ERK1/2)/ERK电泳图;B:p-p38/p38电泳图;C:p-JNK/JNK电泳图
2.6 JNK抑制剂对Aβ1-42诱导的Caspase-3激活的影响 见图5。未经JNK抑制剂处理的SC组、siL-RP2组细胞的Cleaved Caspase-3相对表达与经JNK抑制剂处理的细胞差异无统计学意义(均P>0.05)。

图5 Capase-3激活的电泳图
3 讨 论
AD的主要病理学表现为细胞内大量神经原纤维缠结、老年斑形成以及大量的神经元丢失[4]。Smale等[22]最初用TUNEL染色观察到AD患者脑组织中存在细胞凋亡现象,AD患者海马区TUNEL染色阳性细胞较同龄对照明显增加。许多体内外实验都证实老年斑的主要成分Aβ可以通过激活Caspase-3、引起线粒体功能障碍促进细胞凋亡[23-25]。Caspase-3属于Caspase蛋白家族,是凋亡的终末执行分子。AD患者的神经元中亦检测到激活的Caspase-3和断裂的DNA片段[26]。
近年来,LDLR及其所介导的胞内信号系统在AD发病中的作用日益成为关注的焦点。LRP2作为LDLR家族中最重要的成员,在Aβ代谢中的重要作用逐渐被认识。研究发现,LRP2在脉络膜丛与凝溶胶蛋白相互作用,通过胞吞作用将Aβ经血-脑屏障清除出脑内[27]。同时,具有神经保护作用的类胰岛素生长因子1(IGF-1)、瘦素需在LRP2的介导下才能从外周进入中枢发挥神经保护功能[12-13,15]。本研究小组既往研究发现,LRP2启动子处的多态rs3755166与AD密切相关,rs375166 G>A位点的碱基变异可能影响DNA序列与转录因子的结合活性,导致LRP2表达的相对下降,并使其保护性作用下降继而增加AD的易感性[16],强烈提示LRP2表达下调或功能障碍可能参与AD的发病。
尽管研究显示LRP2在脂蛋白代谢中起到了一定的作用,但目前对于LRP2对神经元的存活是否有直接作用还未明确。为了阐明神经元表达的LRP2在AD发病中的作用,本研究采用Aβ1-42处理SH-SY5Y作为AD细胞模型,以不同浓度Aβ1-42(5 μmol、10 μmol、20 μmol、30 μmol)处理SH-SY5Y细胞24 h。结果显示,Aβ1-42在10 μmol浓度下开始影响SH-SY5Y细胞活力,在20 μmol浓度时SH-SY5Y细胞活力明显下降(52%),因此选择20 μmol作为AD细胞模型处理浓度。与空白对照组相比,Aβ1-42(20 μmol)处理后的AD细胞模型LRP2蛋白表达水平显著降低(P<0.05),提示LRP2可能参与AD的发病机制。本研究发现,siLRP2+Aβ组细胞活力[(40.7±1.9)%]显著低于SC+Aβ组[(54.5±7.5)%](P<0.05),SC+Aβ组细胞凋亡率[(32.4±3.0)%]显著低于siLRP2+Aβ组[(51.8±4.2)%](P<0.01),提示抑制LRP2表达可以使Aβ1-42的细胞毒性增加。
MAPKs是广泛表达的丝氨酸/酪氨酸激酶,是细胞内的一类丝氨酸/苏氨酸蛋白激酶,主要由ERK、JNK和p38信号转导途径组成,可被一系列细胞外的刺激信号激活并介导信号从细胞膜向细胞核传导,与细胞炎症反应[28]、凋亡等密切相关[21]。研究发现,Aβ可以通过激活JNK及p38通路,促进细胞凋亡[29]。Aβ可以通过激活ERK和AKT信号通路参与AD疾病进展[30]。为了明确该途径是否参与抑制LRP2的表达促进Aβ诱导的细胞凋亡过程,本研究在SH-SY5Y细胞系中干扰LRP2后,予以Aβ刺激细胞凋亡,进一步观察ERK、p38以及JNK蛋白磷酸化的变化。结果显示,抑制LRP2表达后JNK磷酸化水平增加,而ERK和p38磷酸化无明显变化。为了明确JNK信号通路是否直接介导了抑制LRP2促进Aβ诱导的细胞凋亡过程,本研究在Aβ刺激前先予以JNK特异性抑制剂SP600125预处理,结果显示,JNK抑制剂不能逆转LRP2抑制导致的Caspase-3激活增加,提示JNK信号通路可能不是直接发挥作用的。研究显示,在海马及皮质神经元中抑制丝氨酸/苏氨酸蛋白激酶(AKT)磷酸化可增强Caspase-3的活化,促进神经元凋亡[31]。本研究小组前期通过免疫荧光和免疫共沉淀发现神经元中LRP2可以与AKT发生相互作用,调控其磷酸化水平参与AD发病发展[32]。有研究发现,AKT信号通路与JNK通路存在交互作用[33],抑制PI3K/AKT信号通路可以促进JNK激活[34]。因此,推测抑制LPR2后对JNK通路的影响可能是受AKT磷酸化水平改变而活化的继发(次要)调节途径。这一推测仍需要在体内外实验中进一步验证。
本研究采用20 μmol Aβ1-42的SH-SY5Y细胞作为AD细胞模型,观察到在AD细胞模型中LRP2蛋白表达下调。通过小干扰RNA抑制LRP2表达后,可以促进Aβ诱导的细胞凋亡。抑制LRP2表达后JNK磷酸化水平增加,ERK和p38磷酸化无明显变化。JNK抑制剂不能逆转LRP2抑制导致的Caspase-3激活增加,提示JNK信号通路可能不是直接发挥作用的。因此,本研究推测抑制LRP2表达促进Aβ诱导的细胞凋亡,其机制可能并不影响MAPKs信号通路,具体机制有待于更深入的研究。
