不同产地当归藤HPLC 指纹图谱
2020-05-12阙祖亮庞丹清陈子隽李金洲魏江存
阙祖亮,庞丹清,陈 勇∗,陈子隽,李金洲,魏江存
(1.广西中医药大学,广西 南宁 530001;2.广西国际壮医医院,广西 南宁 530201)
当归藤为紫金牛科酸藤子属植物当归藤Embelia parvifloraWall.ex A.DC.的根与老藤[1],具有补血活血、强腰膝等功效,常用于治疗血虚诸证、月经不调、腰腿疼痛等[1-3]。当归藤是广西民间地方的传统药物,是壮族瑶族人民在长期与疾病斗争过程中积累起来用于防治疾病的重要药材[4]。
目前国内有关当归藤的报道并不是很多,课题组前期做了当归藤的化学成分预实验[5],并从石油醚部位得到4 个化合物,鉴定其中的2 个化合物为豆甾醇和维生素E[6],同时研究了其中红色素的抗氧化作用[7],运用GC-MS 分析了当归藤挥发油的有关成分[8],在药理作用方面做了毒性、抗炎、镇痛、抗凝血作用的研究[9-10]。迄今对于当归藤药材质量控制的研究仅有单一成分的含有量测定、显微鉴定报道[11-15],为深入开发利用当归藤药材,拟建立其相应的质量控制方法。根据中药多成分、多靶点协同作用的特点,本实验采用HPLC 法对12 批不同产地样品进行测定,初步建立能整体表征中药所含成分及其质量的HPLC 指纹图谱[16-17]。根据HPLC 指纹图谱,共标定12 个共有峰,并鉴定其中的2 种特征峰的成分(儿茶素和没食子酸)。运用IBM SPSS Statistics 22.0 软件及2012 版中药色谱指纹图谱相似度评价系统对12 批不同产地的当归藤药材进行分析,以期为其开发利用提供依据。
1 材料
Thermo U3000 超高效液相色谱仪(美国Thermo Barnstead 公司);Bruke RmicrOTOF-QⅡ型液质联用型高分辨串联质谱仪(德国Bruke 公司)。e2695 型高效液相色谱仪(美国沃特世科技有限公司);BSA224S 电子天平(德国赛多利斯科学仪器有限公司);TG16-WS 离心机(湖南湘仪离心机仪器有限公司);KQ-500DE 型超声波清洗器(昆山市超声仪器有限公司);Simplicity-185 纯水机(德国Millipore 公司)。
乙腈(色谱纯,美国赛默飞世尔科技有限公司);磷酸(西陇化工股份有限公司)。对照品儿茶素(批号110877-201715)、没食子酸(批号110831-201717)均购自中国食品药品检定研究院。水为超纯水,其他试剂均为分析纯。
实验所用药材来源及相关信息见表1,经广西中医药大学药用植物教研室梁子宁副教授鉴定为当归藤Embelia parvifloraWall.ex A.DC.的根与老藤。

表1 样品信息Tab.1 Information of samples
2 方法
2.1 色谱条件 采用Phenomenex C18色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈(A)-0.1%磷酸水溶液(B),梯度洗脱(0~15 min,3%A;15~25 min,8% A;25~60 min,8%~12%A;60~80 min,12% A);体积流量0.8 mL/min;柱温30 ℃;检测波长225 nm;进样量10 μL。
2.2 对照品溶液制备 取儿茶素及没食子酸对照品各适量,精密称定,制成每1 mL 含对照品儿茶素1.012 mg、没食子酸0.416 mg 的贮备液,备用。各取上述贮备液200 μL,置5 mL 量瓶中,定容,制成每1 mL 含儿茶 素40.48 μg、没食子 酸16.64 μg 的对照品溶液。
2.3 供试品溶液制备 称取1.0 g 当归藤药材粉末,过40 目筛,置于50 mL 锥形瓶中,精密加入85% 甲醇20 mL,称定质量,超声处理(功率250 W,频率35 kHz)1 h。放冷,再称定质量,用85%甲醇补足减失质量,摇匀,滤过。将滤液转移至蒸发皿浓缩至干,残渣用85% 甲醇溶解,并转移至2 mL 量瓶中,用85% 甲醇定容,再过0.22 μm 微孔滤膜,即得。
2.4 方法学考察
2.4.1 精密度试验 取广西恭城产地药材(S5),按“2.3”项下方法制备供试品溶液,在“2.1”项色谱条件下连续测定6 次,以6 号峰的保留时间和峰面积为参照,分别统计各共有峰的保留时间和峰面积,测得12 个共有峰相对保留时间与相对峰面积RSD 分别小于1%、3%,表明仪器精密度良好。
2.4.2 重复性试验 称取同一药材粉末6 份,按“2.3”项下方法制备供试品溶液,在“2.1”项色谱条件下,以6 号峰的保留时间和峰面积为参照,测得12 个共有峰相对保留时间与相对峰面积的RSD 分别小于1%、3%,表明该方法重复性良好。
2.4.3 稳定性试验 称取同一药材粉末,按“2.3”项下方法制备供试品溶液,在“2.1”项色谱条件下,分别于0、2、4、8、12、24 h 进样,以6 号峰的保留时间和峰面积为参照,测得12 个共有峰相对保留时间与相对峰面积的RSD 分别小于1%、3%,表明供试品溶液在24 h 内稳定性良好。
2.5 特征峰指认 考虑出峰时间、峰形等因素,并比较样品与对照品的色谱图谱,初步判断1 号峰为没食子酸,6 号峰为儿茶素,将6 号峰确定为特征峰。为进一步确认这2 种成分,采用液质联用方法对当归藤药材中的没食子酸和儿茶素成分进行分析鉴定。
2.5.1 色谱条件 Thermo Gold C18色谱柱(2.1 mm×100 mm,1.8 μm);流动相0.1% 甲酸-水,含20 mmol/L 乙酸铵(A)-乙腈(B),梯度洗脱(0~2 min,10%~30% B;2~6 min,30%~50% B;6~8 min,50%~95% B;8~11 min,95%B;11~11.1 min,95%~10% B;11.1~15 min,10%B);体积流量0.3 mL/min;柱温30 ℃;进样量0.2 μL。
2.5.2 质谱条件 离子源为HESI;辅助气体体积流量10 μL/min;辅助气温度300 ℃;离子传输管温度320 ℃;正离子模式下,鞘气体积流量40 μL/min;喷雾电压3.50 kV;负离子模式下,鞘气体积流量3 μL/min;喷雾电压2.80 kV;m/z扫描范围100~750。
2.5.3 结果分析 图1~5 显示,在相同保留时间处MS 一级二级图谱中的碎片离子,对照品的离子碎片与当归藤样品中相应峰的离子碎片一致。
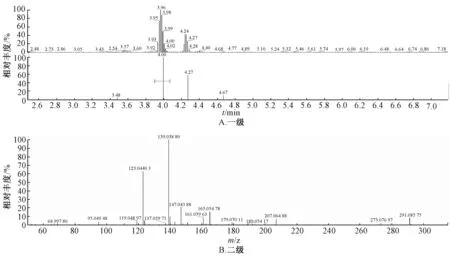
图1 当归藤样品质谱图Fig.1 Mass spectrum of E.parviflora samples
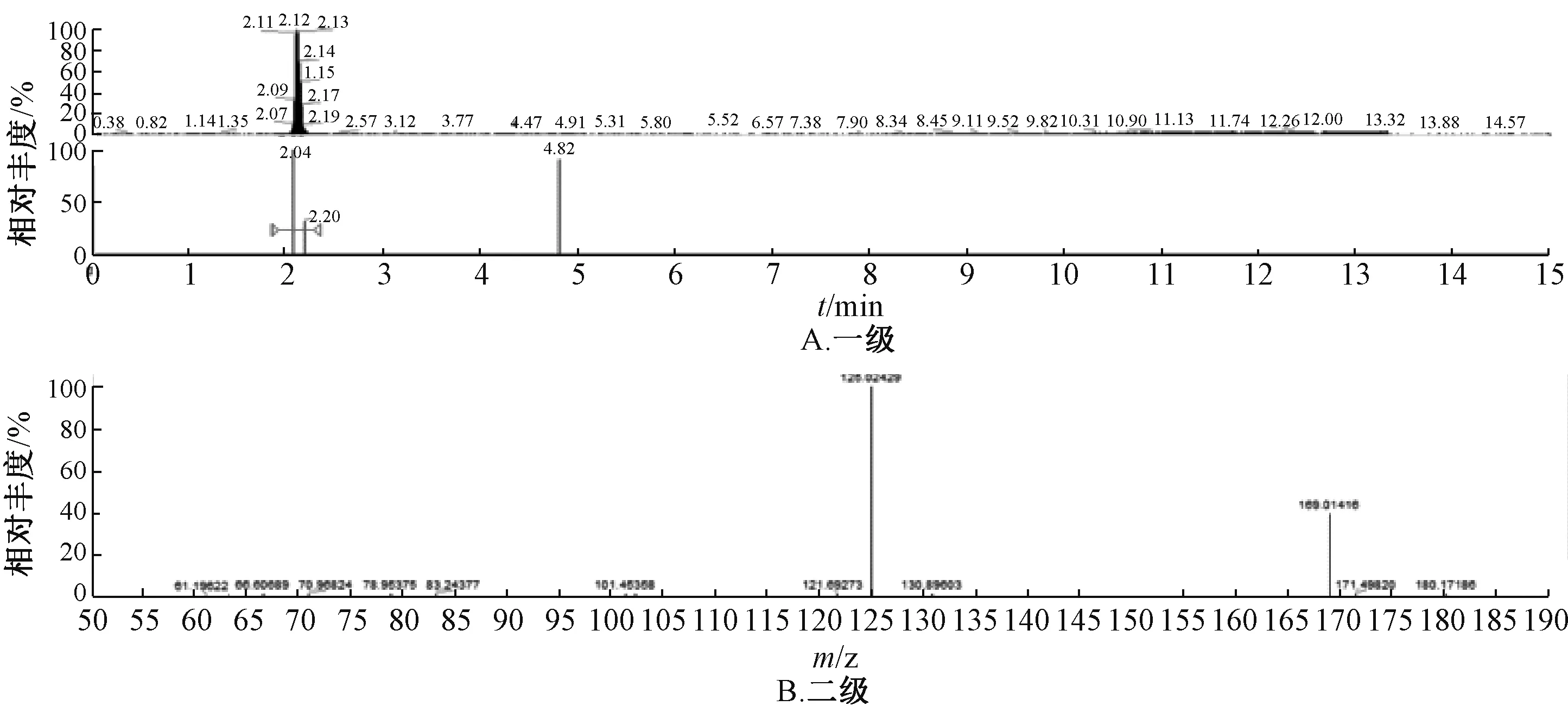
图2 负离子模式下没食子酸样品质谱图Fig.2 Mass spectrum with negative ion mode of gallic acid samples
3 结果
3.1 指纹图谱建立 按“2.3”项下方法制备12批不同产地的当归藤药材供试品溶液,在“2.1”项色谱条件下进样,采集12 批样品色谱图,将其导入2012 版中药色谱指纹图谱相似度评价系统软件,以S5 号的图谱为参照图谱,标定12 个共有峰,并指认1 号峰为没食子酸、6 号峰为儿茶素,以出峰稳定的6 号峰为参照峰,生成当归藤共有模式的指纹图谱,见图6~7。
3.2 相似度分析 将12 批样品的色谱图导入2012 版中药色谱指纹图谱相似度评价系统软件中,选择广西恭城产地当归藤药材作为参照图谱,以中位数作为对照图谱的生成方法,采多点校正,时间窗0.1 min,全峰匹配,建立对照图谱。通过相似度计算结果分析得知,12 批样品指纹图谱与对照图谱的相似度大于0.940,表明各产地药材的化学成分有较好的一致性,但各化学成分含有量存在一定差异。
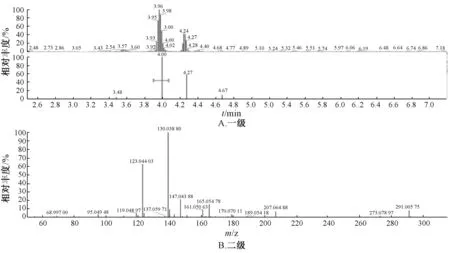
图3 儿茶素样品质谱图Fig.3 Mass spectrum of catechin samples
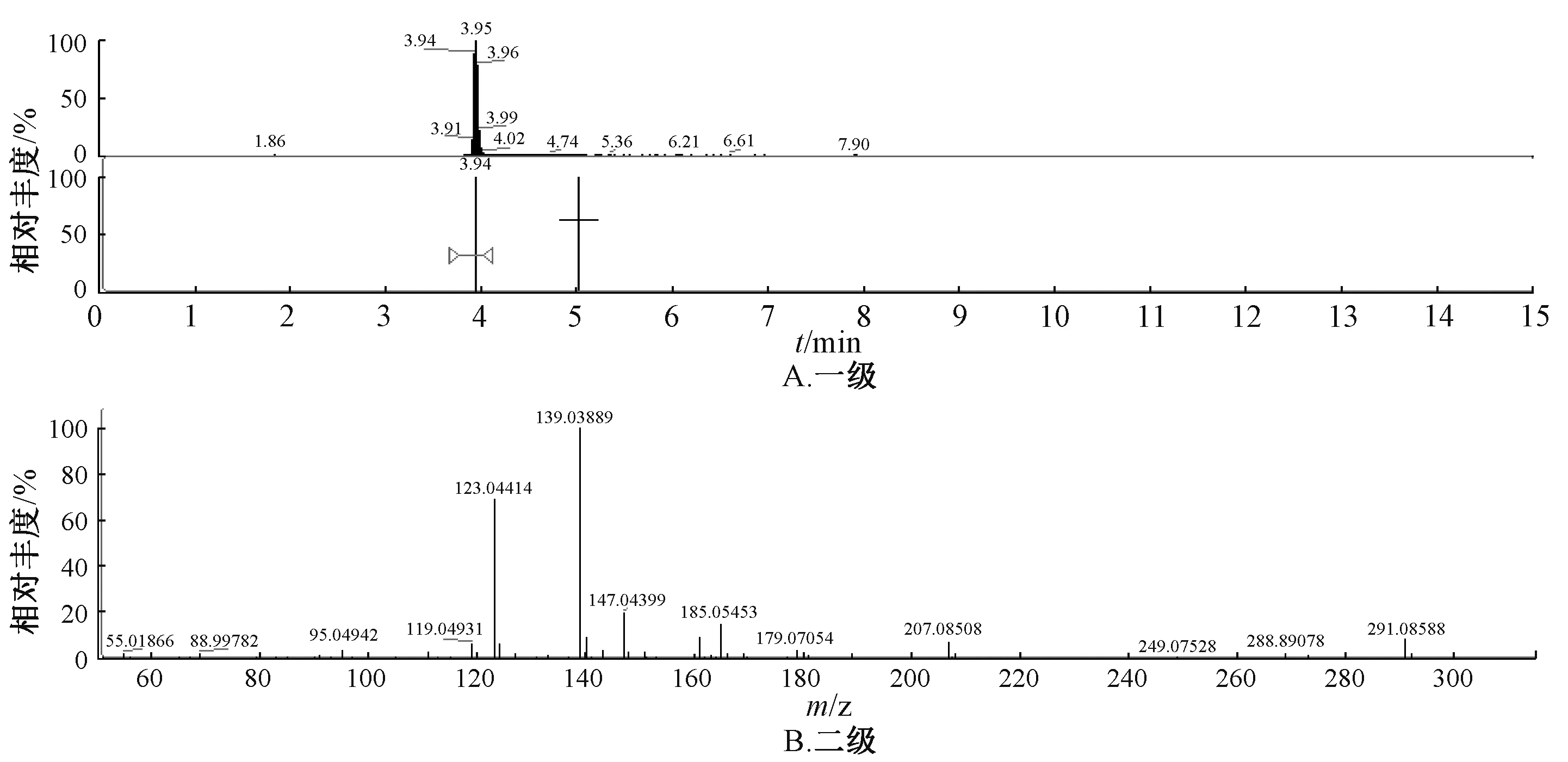
图4 儿茶素对照品质谱图Fig.4 Mass spectrum of catechin reference
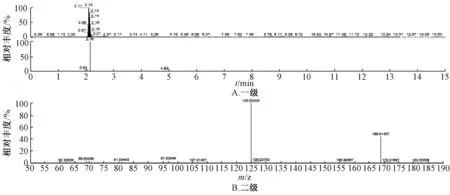
图5 负离子模式没食子酸对照品质谱图Fig.5 Mass spectrum with negative ion mode of gallic acid reference
3.3 共有峰指认 分析各产地色谱图,选择峰形特征明显的色谱作为共有峰,12 批样品样品一共标定出12 个共有峰,选择出峰时间适中且峰形较为稳定的6 号峰儿茶素作为参照峰,分别计算12个共有峰相对于该峰的相对保留时间和相对峰面积,结果见表2~3。
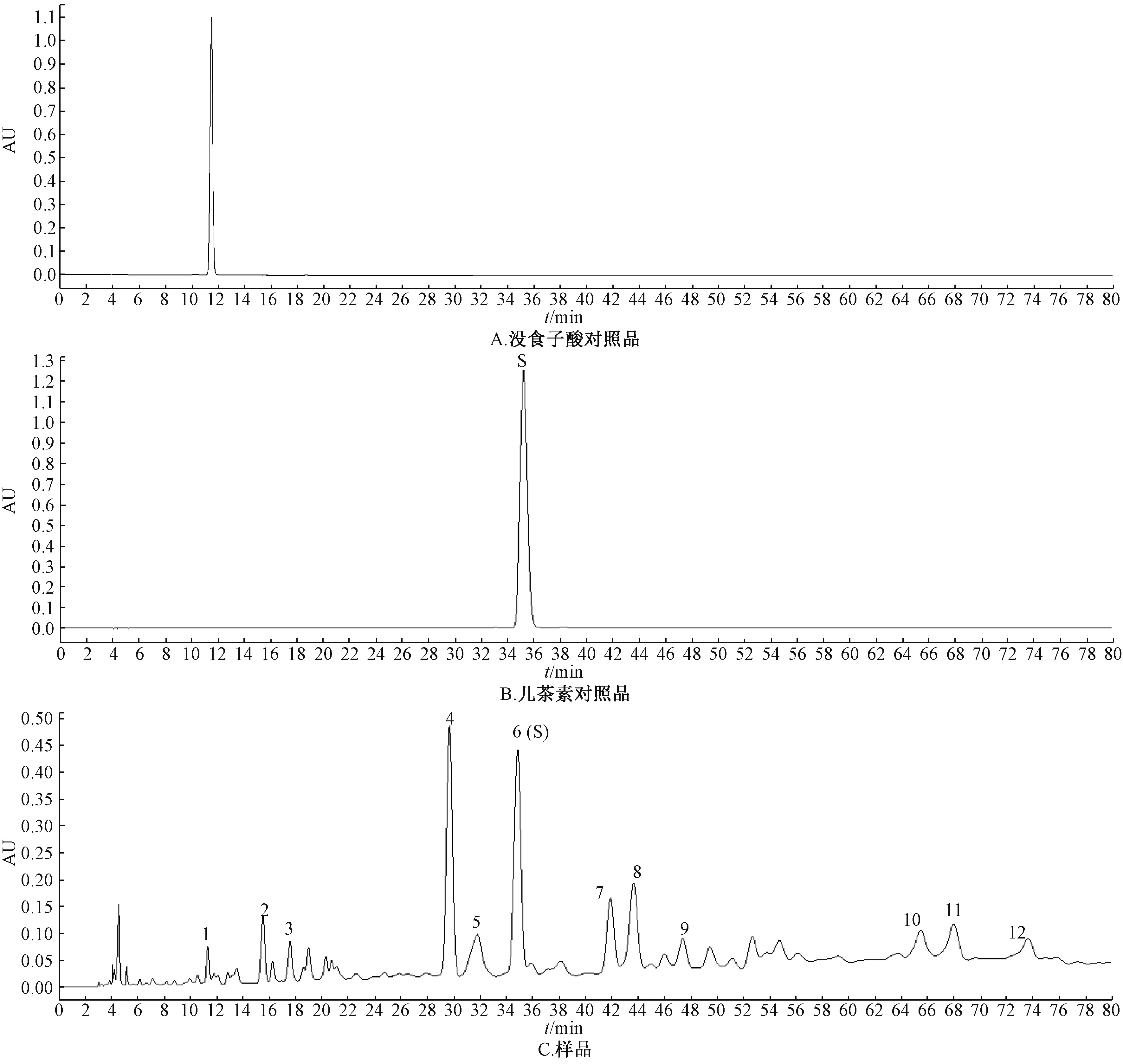
图6 各成分HPLC 色谱图Fig.6 HPLC chromatograms of various constituents
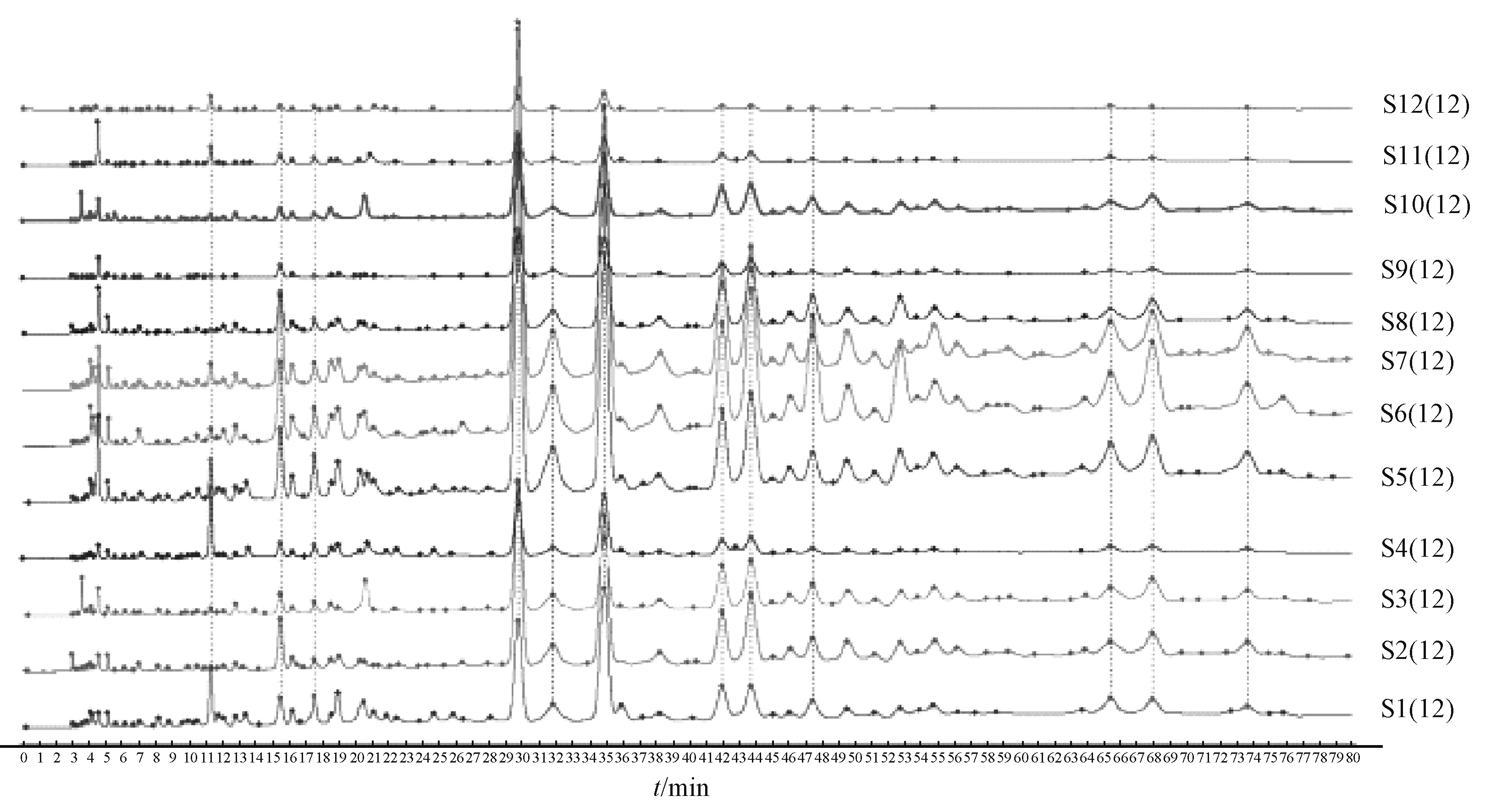
图7 12 批样品HPLC 指纹图谱Fig.7 HPLC fingerprints of twelve batches of samples
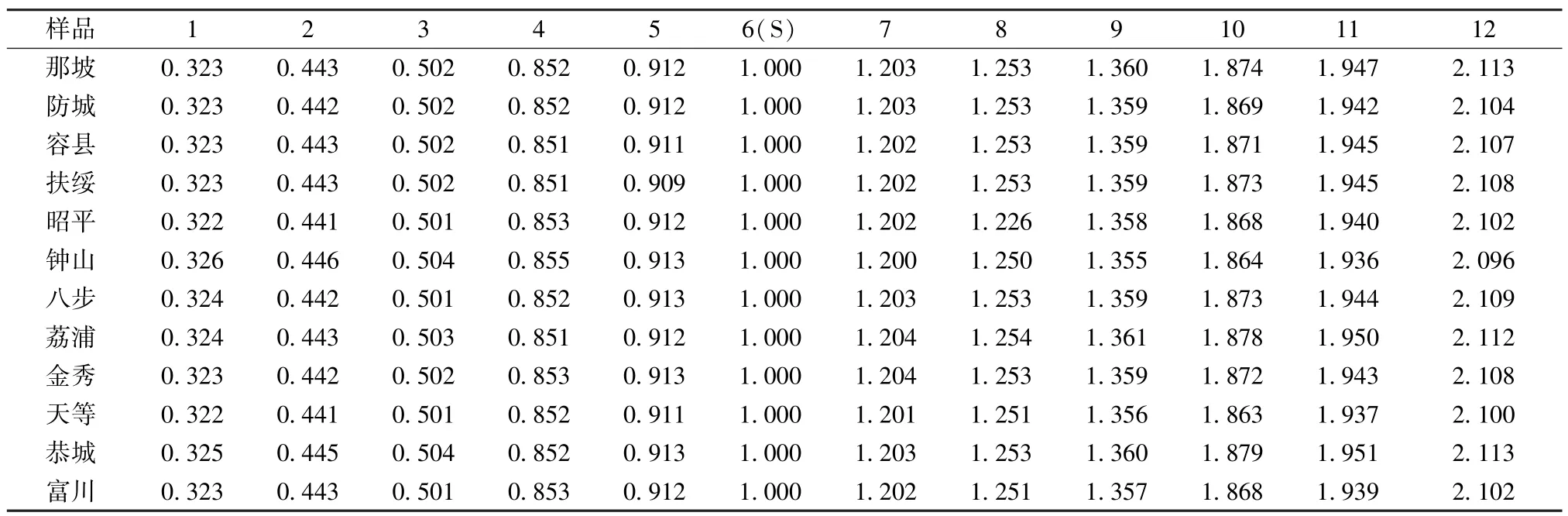
表2 12 批样品共有峰相对保留时间Tab.2 Relative retention time of common peaks of twelve batches of samples
3.4 聚类分析 采用IBM SPSS Statistics 22.0 软件对12 批样品实验数据进行系统聚类分析,采用Ward 法,平方Euclidean 距离(d)为度量标准,结果见图8。表明,当d=10 时,12 批样品可以分为2 类,S5、S6、S7 为一类,其余聚为一类;当d=3 时,后一类又可以聚为2 类,S1、S2、S3、S8 为一类,S4、S9、S10、S11、S12 为一类;由于S5、S6、S7 在地域上差异不大,产地差异较小,故聚为一类。
3.5 主成分分析 采用IBM SPSS Statistics 22.0 软件对12 批样品进行主成分分析,根据特征值与贡献率分析,色谱图中1、2、4、5、6、7、8、10、11、12 号峰的累积贡献率最大,达到95.611%,结果见表5。将对应的特征值做散点图,结合图9、表4 分析,提示这10 个累积贡献率较大的峰可作为评价样品品质的主要因子。通过因子负荷矩阵分析,见表5,得到2 个主成分因子的线性方程。根据主成分1 和2 数据,整理出平面得分图,见图10,从得分图中可以看出这12 批样品分散在不同的区域,由于样品来源于广西的各个市县,表明药材的质量跟地域分布有关。
4 讨论
4.1 色谱柱考察 色谱柱的长短对色谱分离效果有很大影响,本实验考察了150、250 mm 2 种色谱柱,结果表明,后者各色谱峰分离效果较好,故采用250 mm 色谱柱。
4.2 提取方法考察 本实验考察了超声提取和回流提取2 种方法,并考察了不同体积分数甲醇和乙醇,通过比较出峰数、峰形等,选择85% 甲醇超声60 min。
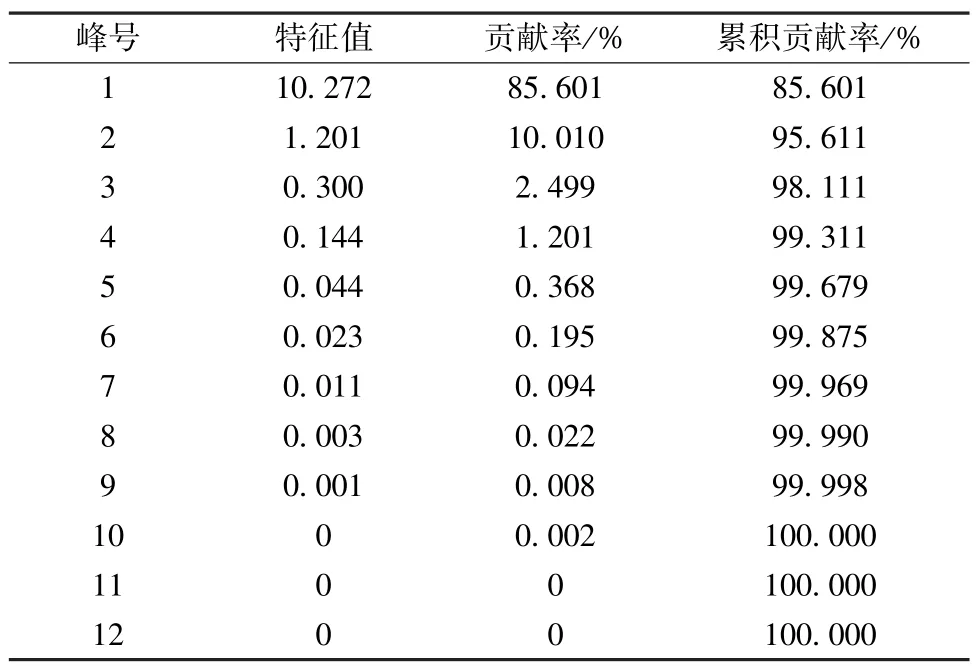
表4 主成分初始特征值和贡献率Tab.4 Initial eigenvalues and contribution rates of principal components
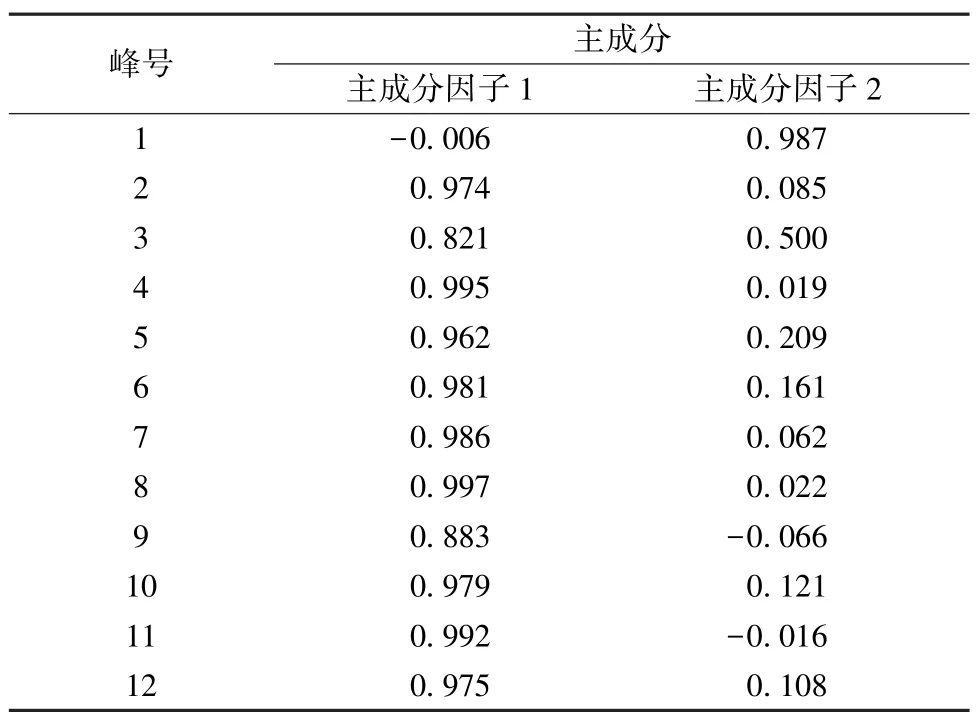
表5 因子负荷矩阵Tab.5 Matrix of factor loadings
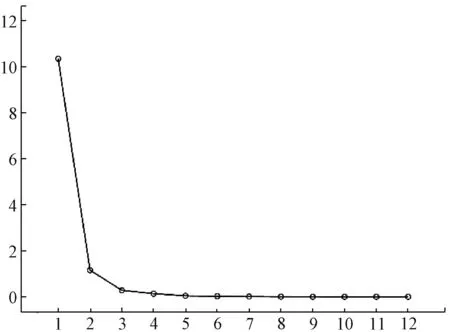
图9 各成分主成分分析散点图Fig.9 Scatter diagram of principal component analysis of various constituents
4.3 色谱柱以及流动相考察 本实验考察了3 种不同型号的色谱柱,Phenomenex C18、Inertsil C18和Thermo C18,并同时考察了4 种常见的不同流动相体系,根据峰形,峰数量以及分离度等综合考虑,以Phenomenex C18色谱柱、乙腈-0.1%磷酸梯度洗脱时,峰数较多,峰形较好。
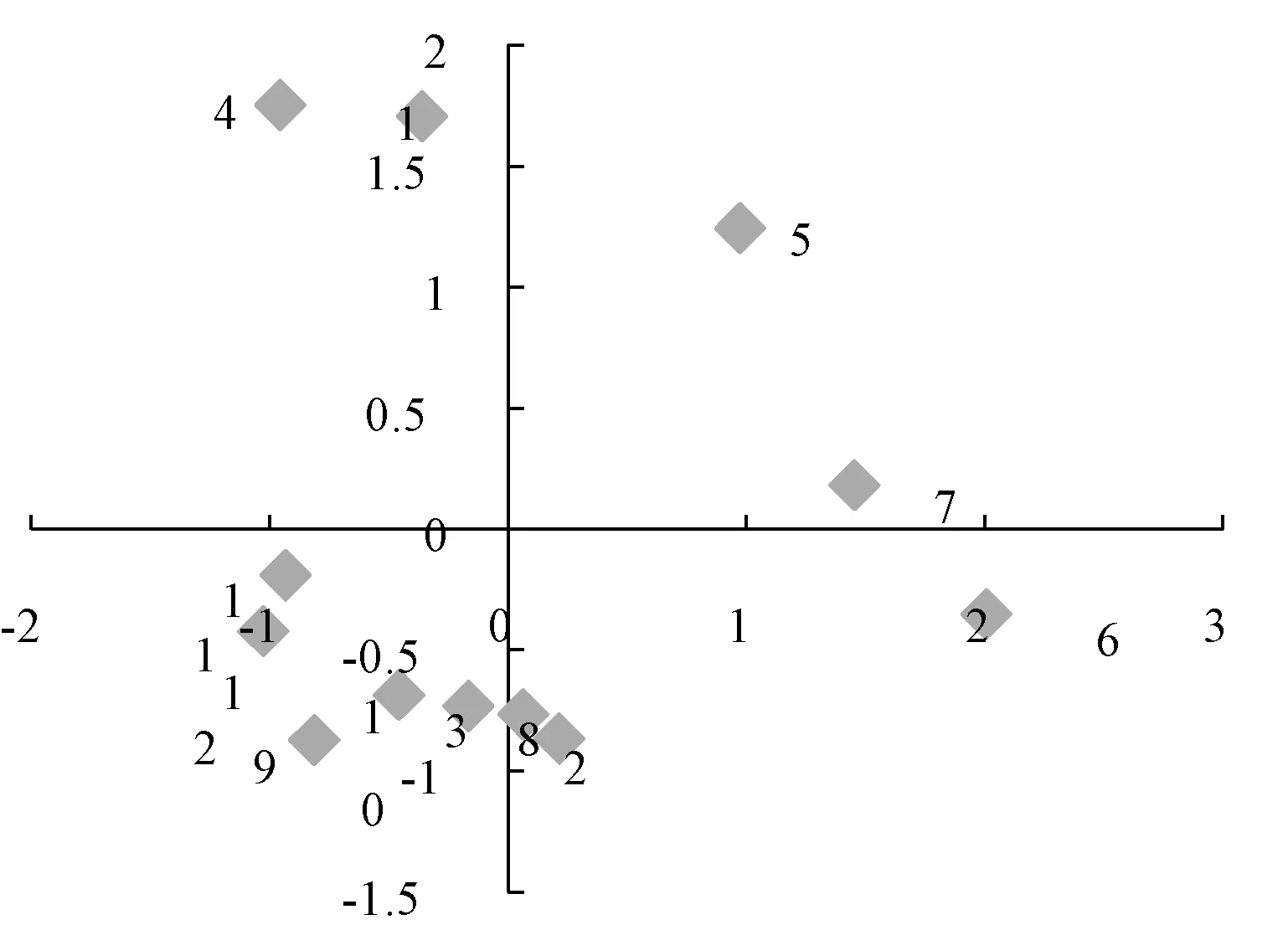
图10 主成分平面得分图Fig.10 Score graph of principal components
4.3 波长考察 本实验采用PDA 检测器进行全波长扫描,结果表明在225 nm 波长下,色谱图的信息丰富,各色谱峰分离效果较好,无明显拖尾现象,基线相对平稳,故检测波长为225 nm。
4.4 柱温、体积流量考察 对比3 种不同体积流量的色谱图,结果表明0.8 mL/min 的出峰时间较适宜,且峰分离度较好,故体积流量为0.8 mL/min。在柱温选择方面,对比了4 种常见的柱温,以30 ℃时色谱峰分离效果、峰形等较好,故柱温为30 ℃。
5 小结
本实验采用HPLC 指纹图谱结合特征峰含有量的变化控制当归藤药材质量,比单纯测定药材的某一成分含有量控制药材的质量更全面客观;聚类分析结合HPLC 指纹图谱,方法稳定高效、简便且专属性强,以期为对进一步研究当归藤质量评价及药效物质基础提供参考。
