美国FDA 对公共卫生紧急事件的防范和应对措施
2020-05-10陈谨徐然赵东杜涛
陈谨,徐然,赵东,杜涛
(汉佛莱医药顾问有限公司,马里兰州 柏赛斯塔 20817)
在过去的30 年中,全球的经济、政治、自然环境都发生了巨大的变化,也对公共卫生领域提出了前所未有的挑战。1984 年当美国国立过敏和传染病研究所所长弗奇博士在世界地图上标示传染性疾病的发生时,全球仅有艾滋病(AIDS)这一种新发传染性疾病。直至2017 年,这张地图已经布满大大小小的传染性疾病的爆发点,其中包括中东呼吸综合征(MERS)、高致病性禽流感(HPA2)、寨卡病毒病(ZVD)、埃博拉出血热(Ebola hemorrhagic fever)、H7N9 禽流感、甲型H1N1 流感,以及严重急性呼吸综合征(SARS)等等[1]。每一种疾病都来势汹汹,人类又束手无策。另一方面,受全球气候变暖影响,各类自然灾害给公共卫生领域带来诸多挑战,生物、化学、放射性/核方面的恐怖袭击威胁也成为公共卫生考虑的重要议题。面对日益增加的各类公共卫生威胁,美国政府非常重视对此类事件的防范和应对。早在克林顿政府时期,美国就开始了立法程序,在法律和政策上为相关政府职能机构提供法理和政策支持,同时也规范各职能部门在公共卫生紧急事件中的角色和工作内容,协同政府和社会各界的资源整合。
应对公共卫生威胁是一个集危机管理、紧急事件防范和应对、资源整合、人员培训等诸多因素的系统工程,其中医疗应对产品(medical countermeasure,MCM)的使用则处于这一工程的核心地位。美国FDA 是美国卫生和公众服务部(HHS)直属机构,在应对公共卫生紧急事件中,担负着保障MCM 的安全性、有效性和可靠性的重要职能,保护美国公众免受公共卫生威胁。本文从MCM 的角度,简介FDA 对公共卫生紧急事件的防范和应对措施,以期对我国相关产品的评审、监管和使用有所借鉴。
MCM 是由FDA 监管的医疗产品,包括生物制品、药品和医疗器械,用于应对恐怖袭击引起的生物、化学、放射性/核威胁或自然发生的新发疾病引起的公共卫生威胁,该类产品主要用于诊断、预防、保护或治疗由上述威胁引起的疾病。
1 医疗应对产品的相关立法
早在20 世纪90 年代,美国政府开始通过立法程序来发展和储备应对生物威胁的MCM。2001 年“9·11”恐怖袭击之后,美国政府加快了应对公共卫生紧急事件的相关立法,这些立法的核心都与促进MCM 的研发、评审、授权使用相关,以期可以防范、应对公共卫生紧急事件。表1 对这些相关法律进行了简单的梳理。
2 医疗应对产品激励政策
在上述一系列法律框架下,FDA 制定了相关的审评审批机制、内部流程和技术指南,以促进MCM 的研发,加快审批程序,以期相关产品在公共卫生紧急状态下可以被迅速提供给公众。FDA 支撑上述目标的三大策略是:①给予MCM 以及相关技术最高程度的评审优先权;②推动评审MCM 的监管科学进步;③为有效对公共卫生需求作出反应,进一步完善技术指南并加强与开发者的沟通。
2.1 提升评审优先权
FDA 在医疗产品的评审中有4 种快速评审程序,分别是快速通道、优先评审、加速批准和突破性疗法。MCM 在评审过程中可以使用这些快速评审程序。另外,针对MCM,FDA 也有一些特别条款。其中,值得注意的是根据2016 年《21 世纪治愈法案》,FDA 建立了一个新的优先评审机制。这个被称为“重大威胁MCM 优先评审”的政策,允许FDA 将优先评审券授予符合要求的应对重大威胁的MCM[2]。一旦FDA 作出优先评审的认定,FDA 将在6 个月内完成产品的上市申请评估,而通常的标准评审时间为10 个月。此外,该产品的申请人可以将这一优先评审券使用于另一个未获得优先评审的待审产品。此优先评审券可以通过市场进行转让,目前优先评审券市场价值在1 亿美元左右。FDA 期望评审优先权的提升、评审时间的缩短以及优先评审券的增值,可以鼓励更多此类产品的开发。
2.2 监管科学的发展
MCM 在开发过程中常伴随出现一些独特、复杂的监管科学挑战。例如生物、化学、放射性/核威胁这一类高危威胁并不会自然发生,应对此类威胁的医疗产品因为种种原因无法进行人体实验,无法直接证明其有效性。鉴于这种不同于传统医疗产品的情况,FDA 近年来一直在执行MCM 倡导计划(MCM Initiative Program,MCMi),目的在于通过发展新的工具、标准和方法来评估MCM 的安全性、有效性、质量和性能,帮助将尖端科技转化为安全有效的MCM。在此计划的框架内,FDA 建立了广泛稳固的内部和外部合作,主要的研究领域如下:①识别、发展和鉴定MCM,开发新型的工具来评估安全性和有效性;②验证下一代基因测序体外诊断试剂盒;③开发参照品;④评估产品性能、设计和应急医疗设备(包括个人防护设备)的重复使用;⑤增强应急防范和响应能力;⑥提高MCM 生产能力。
2.3 技术指南的更新和科学透明的交流沟通
FDA 十分重视技术指南的更新和沟通的公开透明,为MCM 在内的医疗产品提供合适的法规意见和支持。2018 财年,FDA 推出或更新了42 个涉及MCM 的指南文件。这些文件中不仅有直接指导MCM 开发的指南,也有促进MCM 开发和可用性的指南。FDA 也建立了正式会议机制,MCM 申请人可以通过这项机制与FDA 进行直接交流,咨询监管机构在产品开发中的意见。根据FDA 的报告,2018 年度FDA 在三大中心与MCM 申请人召开了超过100次的会议。在指南和会议之外,FDA 也通过其他渠道和产品申请人进行沟通,如咨询委员会会议、在线教程等。FDA 的积极行动都将鼓励MCM 的开发。
在一系列产品激励政策的行动下,FDA 成绩斐然,仅在2018 财年,FDA 一共批准了28 个用于MCM,其中9 个药品/生物制品,19 个医疗器械(包括体外诊断试剂盒)。
以用于治疗天花的药品TPOXX 为例,世界卫生组织(WHO)虽然在1980 年即宣布天花已经绝迹,但是长久以来各国政府仍然担心天花病毒被作为生物武器使用,而由于开发相关药物的成本较高,潜在经济收益又非常有限,企业研发动力不足,美国政府通过一系列评审激励鼓励研究。2018 年SIGA Technologies Inc 的产品TPOXX 获批上市。在整个评审过程中,该药成功获得了快速通道和优先评审,整个新药申请(NDA)的评审时间仅为7 个月左右,并且还获得了重大威胁MCM 优先评审券[3]。2018年11 月,SIGA 即以8 000 万美元的价格将此评审券出售给礼来公司。此外,TPOXX还被授予孤儿药认证,因此在税收和其他方面TPOXX 也可享受一些优惠。
另外一个产品,针对埃博拉病毒的疫苗Ervebo在2019 年12 月获批。该疫苗由美国默克公司研发,评审过程中不仅获得了“热带病优先评审券”,还获得了突破性疗法认定。这些快速评审程序的使用,使得FDA 和默克在研发和评审过程紧密合作,整个评审时间不到6 个月[4]。
3 医疗应对产品紧急授权使用
公共卫生重大威胁出现时,FDA 也肩负着将MCM 快速推向公众、发挥作用的职责。以下将从法律、法规和几次重大疫情中FDA 紧急授权使用MCM 的案例这几个方面进行介绍。
3.1 法案解读
3.1.1 《联邦食品、药品和化妆品法案》第564 节
FD&C Act 第564 节由2004 年《生物盾牌计划法案》修订而成,并由2013 年《大流行和全方位灾害准备再授权法案》、2016 年《21 世纪治愈法案》和2017 年《公共法115-92》进一步修订而成。
FD&C Act 第564 节规定了EUA 的要求:①确认存在紧急情况;②宣布进入紧急状态;③符合FDA 的规定标准[5]。确认紧急状态,实施EUA 必须基于如下几种情况之一:①国土安全部部长分析认定国内出现国内紧急状态,或者出现紧急状态的可能性很大,生物、化学、放射性/核袭击风险增加;②国防部(DoD)部长分析认定出现军事紧急状态,或者出现紧急军事状态的可能性很大,生物、化学、放射性/核袭击的风险增加;③卫生部长分析认定国内将处于或正处于根据美国《公共卫生服务法案》(Public Health Service Act, PHS Act)第247d 条所包含的那些影响国家安全的公共卫生紧急状态,包括受到特定1 种或多种生物、化学、放射性/核袭击的风险增加,或出现这些物质引起的特定疾病和状况。
如果紧急状态已经确认存在,基于FD&C Act第564 节,美国卫生和公众服务部部长可以声明EUA的使用[4]。FDA被要求公布每一项EUA的通告。在决定宣布紧急状态时,美国卫生与公众服务部部长审查提交的EUA 信息,并可咨询EUA 工作组的联邦官员[6]。一个紧急声明可以支持多个EUA[7]。
当美国卫生和公众服务部部长宣布紧急状态的EUA 使用声明后,FDA 专员与国立卫生研究院(NIH)和疾病控制和预防中心(CDC)商讨(根据紧急情况而决定是否可行),并根据会议法规和其他标准签发EUA(见图1)。
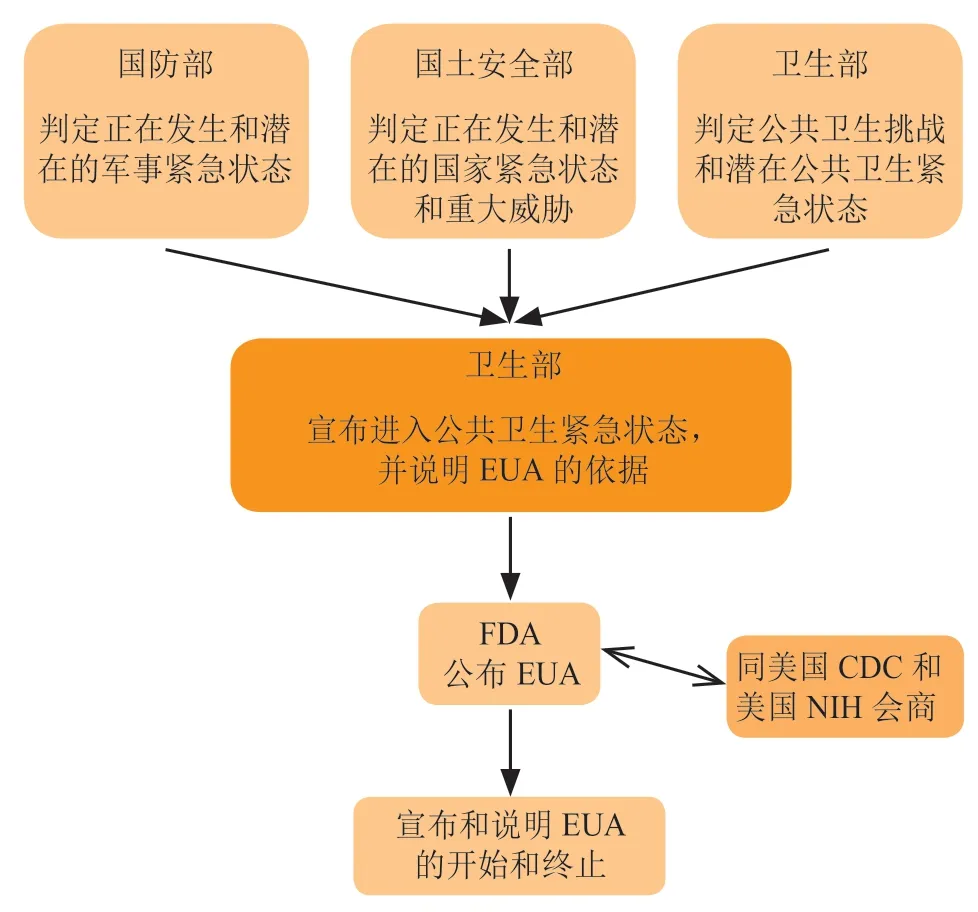
图 1 紧急使用授权的操作程序Figure 1 Operation procedures of EUA
3.1.2 《医药产品紧急使用授权指南》
美国医疗产品EUA 由美国国会于2004 年建立,作为《生物盾牌计划法案》的一部分,通过对FD&C Act 进行修订,增加第564 节,宣布当面对与美国公众或军队受到攻击的风险加重有关的紧急状态时,FDA 局长可以授权使用未经批准的医药产品或已获批医药产品用于未经批准的用途。
FDA 于2007 年7 月公布《医药产品紧急使用授权指南》(Emergency Use Authorization of Medical Products and Related Authorities),建立了EUA 的操作程序[8]。在应对2009 年甲型H1N1 流感之前,FDA 只发布过2 个医药产品EUA,其一是用于预防吸入性炭疽热的药品,其二是2008 年发布的用于邮寄模式的抗生素紧急使用套装。2016 年颁布的《21世纪治愈法案》和2013 年PAHPRA 修订了FD&C Act 第564 节,进一步完善了EUA。
在FDA 的内部组织结构,由局长办公室和首席科学家作为主导,协调反恐和新发威胁办公室、生物制品评审和研究中心(CBER)、药品评审和研究中心(CDER)以及设备仪器与放射健康中心(CDRH)共同应对紧急状态。FDA 局长在与NIH 和CDC 讨论后,由FDA 最终发布EUA。
EUA 不要求医药产品位于特定的研发阶段。已上市的药品未经批准相关适应证,未上市药品处于临床试验阶段或完成临床试验后的NDA 阶段均可申请EUA。潜在的EUA 产品包括药品、生物制品(如疫苗、血液制品和生物疗法)和器械(如体外诊断试剂)。
FDA 会根据多种因素来建立申请的审查优先级,其中包括:临床疾病或状况的严重性和发生率;产品的公共卫生需求以及其他潜在MCM 的安全性和有效性;急需治疗的紧急情况;有关产品在预防、治疗或诊断疾病方面安全有效信息的可用性和充分性;具有能够确保国家的安全的潜在作用。
EUA 的申请人首先应该通过pre-EUA 的途径与FDA 进行所需数据和信息的沟通,并通过EUA递交的形式提交其正式请求,包括参考FDA 先前已审查过的Pre-EUA 申请,并要求通过相同程序签发EUA。在卫生部根据FD&C Act 第564 条发布EUA声明之前,申请者不应提交正式的EUA 正式请求。如果FDA 以防备为目的确需签发EUA,也需由卫生部评估并授权。FDA 通常在整个EUA 流程中都与卫生部和其他相关政府部门密切协调,包括需要发布EUA 声明以及与联邦政府组织间就EUA 进行任何协商[5]。通常情况下,正式签发EUA 的申请会从Pre-EUA 的FDA 反馈中受益。
EUA 的申请人应通过EUA 递交的形式提交其正式请求,包括参考FDA 先前已审查过Pre-EUA申请,并要求通过相同程序签发EUA。通常,EUA提交中的数据和其他信息在此之前应该已经由FDA通过Pre-EUA 审阅,并受益于FDA 与申请者在Pre-EUA 过程中的沟通和反馈。
如果情况允许,且通过Pre-EUA 的互动可以提供足够的信息供FDA 事先审核,则FDA 会迅速发布EUA。通常,FDA将视具体情况,并根据产品介绍、产品之前的申请状态、紧急或潜在紧急状态的性质、申请EUA 的机构和组织以及当前的工作量来确定审查和下一步行动的时间表。
FDA 局长(或其指定人员)将签署授权MCM的正式信函,其中包括授权产品及其用途的说明、该产品的任何禁忌证、授权的标准、授权范围、豁免某些要求(如果适用)以及授权使用的任何条件。授权的EUA 将由签署的授权书和任何随附的授权材料(例如医疗保健专业人员的情况说明书、接收者的情况说明书、使用说明等)组成[9]。
FDA 通常期望EUA 产品按照cGMP 标准进行合规生产、存储和分发;但是,在考虑到具体情况和任何其他提议的方法后,可以根据具体情况在EUA 中授予限制或豁免[5]。
在紧急情况下,FDA 可以在适当的范围内放弃部分分发要求。例如,针对大规模紧急响应的操作,为了尽快分发MCM 以保护公众健康,可能会要求大量人员在集中位置或非传统卫生保健设置的位置(通常称为“分配点”)接收医疗产品。在这种情况下,要求每个人在接受EUA 授权的产品之前与持执照的执业者进行配合可能不切实际。
FDA 可能会基于触发EUA 的生物、化学、放射性/核威胁紧急情况而豁免其适用的风险评估和缓解策略(REMS)要求。如果确定豁免,则该豁免可适用于所有REMS 要素。
当EUA 声明被终止后,基于该声明发布的所有EUA 将不再有效。EUA 声明于以下日期中的较早者终止:美国卫生和公众服务部认为促成该声明的情况已经终止或是产品的批准状态改变,使产品的授权使用不再是未获批状态。
3.1.3 《试验用药品扩大使用指南》
1987 年,美国FDA 建立研究用药品扩大使用的监管途径,允许重症或晚期患者在临床试验之外有机会获得研究性药品以治疗疾病。2009 年8 月,美国FDA 进一步对法规进行了修订,明确了研究用药扩大使用的规范流程。2016 年6 月,美国FDA颁布了相关指南文件:《以治疗为目的的试验用药品同情使用相关问题解答指南》(Expanded Access to Investigational Drugs for Treatment Use: Questions and Answers Guidance for Industry)、《单个患者拓展使用申请》(Individual Patient Expanded Access Applications)和《有关试验用药品收费的相关问题指南》(Charging for Investigational Drugs under an IND: Questions and Answers),指南中对研究用药品扩大使用进行了详细的解释和说明。2016年12月,美国正式通过了《21 世纪治愈法案》,修订了FD&C Act,新增第561A 条“研究性药品的拓展使用要求”条款。
根据美国2009 年发布的 《试验用药品扩大使用指南》,FDA 扩大使用程序(也称特许使用)是经医生提出申请,由FDA 授权批准后,可以在某些特定的病人的治疗中使用。且法规指出,如果在提交书面申请前,需对患者进行紧急治疗,FDA 允许在未提交书面申请的情况下扩大药物使用。FDA 审评员可通过电话允许紧急情况下使用药物。
当满足以下所有条件时,医生可能决定通过个例患者紧急研究用新药(EIND)申请(见表2)来请求使用某款研究用产品:①患者患有严重的疾病或状况,或者其疾病或状况立即威胁生命;②没有替代疗法或产品来诊断、监测或治疗该疾病或状况;③无法将患者纳入临床试验;④潜在的患者利益大于治疗的潜在风险;⑤提供研究用医疗产品不会干扰可以用于支持医疗产品开发或销售该治疗适应证的研究试验。

表 2 紧急研究用新药申请递交时间表Table 2 Submission timeline of EIND
表3 详细地对比了EUA 产品、EIND 产品和一般情况下IND 产品和已上市药品的相关标准[10]。EUA 和EIND 都是在紧急情况下的授权或批准,但两者在适用情形和使用范围上有明显区分。其中EUA 是在实际的或潜在的紧急状态下对未获批准的医药产品的使用及已获批准产品的未获批准用途的授权,而EIND 主要的适用情形主要为有严重疾病或立即危及生命的单一病人。因此,在紧急状态下,如疾病爆发时,EUA 和EIND 会在不同适用范围下对疫情的爆发有所缓解。而在正常状态下,单一病人同样可以通过EIND 授权而获益。对于处方药安全报告的要求、知情同意的签署与否以及机构审查委员会的申请时限,EUA 会有更宽泛的条件,而EIND 则需要遵守基本的IND 法规要求[10]。
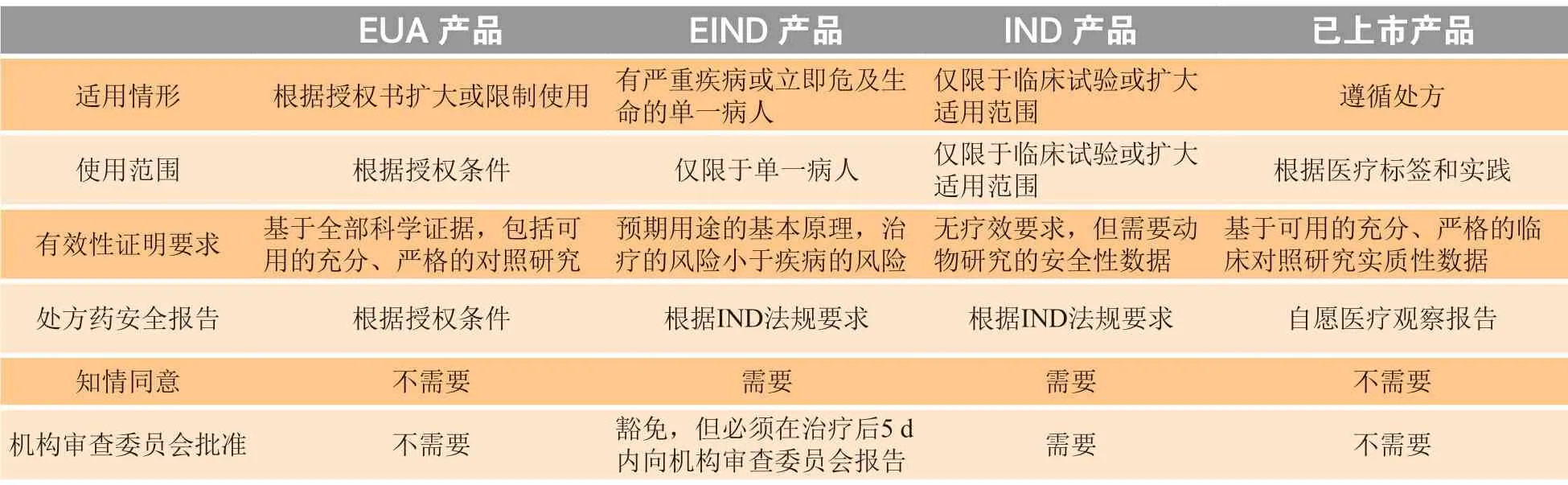
表 3 EUA 产品、EIND 产品、IND 产品与已上市产品的相关标准比较Table 3 Comparison of criteria between EUA, EIND and IND products and those on the market
3.2 案例分析
在2009—2010 年应对甲型H1N1 流感期间,FDA 发布了23 个产品的EUA,其中包括3 个抗病毒药品,1 个个人呼吸防护装置(N95 防护口罩)和19 个病毒诊断试剂。其中,3 个抗病毒药品包括1 个未获批准药品帕拉米韦静脉注射液和2 个已批准药品但未获批准用途的磷酸奥司他韦(达菲)和扎米那韦。
磷酸奥司他韦是一种作用于神经氨酸酶的特异性抑制剂,抑制流感病毒在人体内的传播以起到治疗流行性感冒的作用。达菲于1999 年3 月递交NDA 并于同年10 月获得上市许可,批准的适应证为出现症状不超过2 d 的成人流感,此外还被批准治疗1 岁以上流感症状不超过2 d 的儿童流感患者,且可以用于预防成人和年龄在13 岁及以上的青少年流感。达菲于2001 年10 月在中国获批准上市。2009 年H1N1 流感爆发,FDA 在该年10 月给予达菲EUA,扩大其适应证范围,将达菲用于1 周岁以下患病儿童直至2010 年6 月EUA 结束。同年,达菲进入WHO,并作为抗流感必需药物列入“基本药物”名单中,推荐各国按照该国人口的1/4 进行药品储备。在甲型H1N1 流感的EUA 结束后的第3年,即2012 年12 月,FDA 批准扩大达菲适应证,允许其用于流感症状不超过2 d 的2 周以上儿童的治疗。
帕拉米韦注射剂是由BioCryst Pharmaceuticals开发的用于治疗流感的抗病毒产品。该产品也是一种神经氨酸酶抑制剂,可单剂量静脉或肌肉注射,用于治疗急性或单纯性流感;也可多剂量静脉注射治疗重症流感。2009 年甲型H1N1 流感大流行期间,帕拉米韦仍在开发阶段,尚未获得FDA 上市许可。BioCryst 在2008—2009 年间完成了Ⅱ期临床试验,但结果显示该药对季节性流感效果不佳。与此同时,数项Ⅲ期临床试验正在甲型H1N1 流感大流行的东亚地区开展。2009 年4 月26 日,美国卫生部因甲型H1N1 流感大流行,宣布进入紧急状态。2009 年10 月有报道显示,8 位住院重症病人接受了帕拉米韦注射治疗,效果良好。2009 年10 月23 日,FDA授予帕拉米韦EUA。EUA 发布以前,临床试验以外的病患只能通过EIND 使用帕拉米韦,而这种使用方式取决于厂商的意愿以及各方受理这种申请的能力。病人获取该治疗方案的效率不高。EUA 的发布使得全美范围内因甲型H1N1 流感住院患者可以便捷地获得此项治疗。EUA 发布之后,帕拉米韦使用直线攀升,其使用量是EUA发布前的38 ~ 48倍[11]。进入2010 年,疫情逐渐消退,2010 年6 月23 日,美国卫生部宣布紧急状态结束,帕拉米韦的EUA 也相应过期,临床试验以外的患者不能再使用该药。在2009 年10 月23 日至2010 年6 月23 日的8 个月中,1 274 名住院病人通过EUA 的方式接受了帕拉米韦静脉注射治疗,其中很多为危重病人,41%的病患需机械通气[12]。虽然,在EUA 的使用中无法对帕拉米韦的安全性和有效性作充分评估,但其在甲型H1N1流感大流行期间提供了可能的治疗手段,为挽救生命、保护公众健康起到了积极作用。同时,这也是FDA 历史上首次将EUA 授予一个未获得上市批准的研究用药,对FDA 而言,在紧急状况下的应急行动也是一次有益操练,提升了监管机构在紧急状态下的反应速度。整个疫情期间,美国卫生部采购了11 200 份供成年患者5 d 注射疗程的药品。
通过应对2009 年的甲型H1N1 流感,FDA 在各个方面取得了宝贵的经验,包括通过药品有效期延长计划(SLEP)缓解药品短缺的问题。SLEP 延长了符合条件的药品和其他联邦储备物资的有效期,该计划由美国DoD 与FDA 合作管理[13]。其次,扩展已获批药品的适应证以及使用未获批的药品用来治疗新型疾病以避免其爆发。另外,FDA 和CDC在应对甲型H1N1流感的紧急事件中实现信息公开,将所有发布的EUA 公开供公众阅览。
4 医疗应对产品的安全性和有效性评估
在公共卫生紧急状态时,及时推出MCM 对降低疫情中的患病率和死亡率至关重要。另一方面,很多MCM 可能是第一次使用,也可能仅适用于当下紧急状况,而产品本身又很可能处于开发早期阶段,人们对其知之不多。同时,紧急状态下受各种因素的限制,人们无法对所知不多的MCM 进行详尽评估。因此,MCM 的安全性和有效性评估不同于常规医疗产品。表4 对比了公共卫生紧急状态和常规研发状态时医疗产品评估的异同[14]。
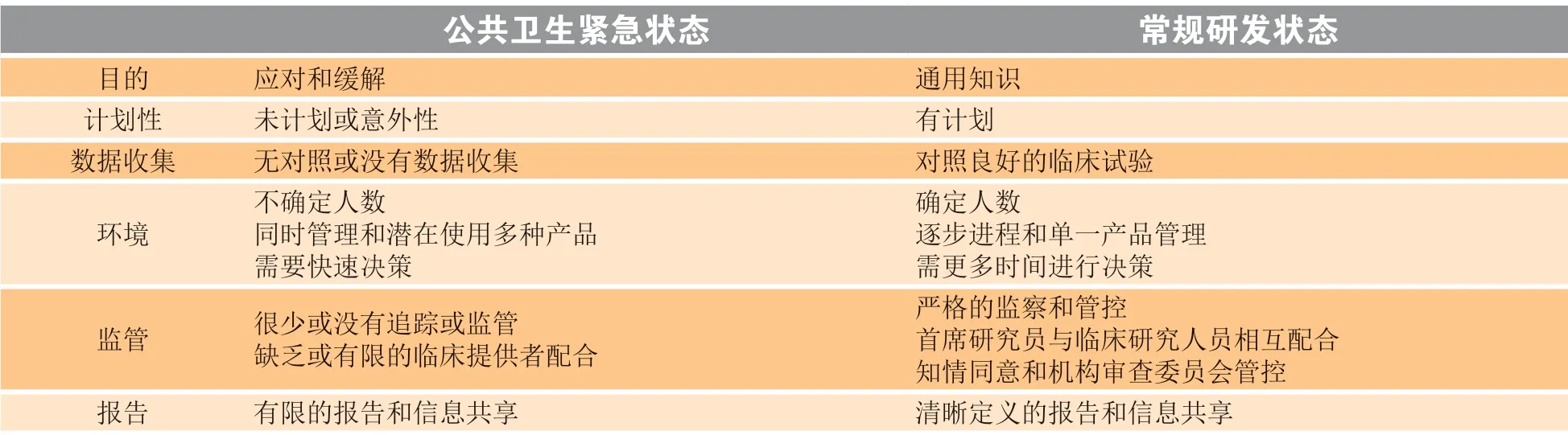
表 4 公共卫生紧急状态和常规研发状态时医疗产品评估的比较Table 4 Comparison of medical product review under public health emergency and conventional R&D
一般而言,FDA 通常会对医疗产品是否可以上市进行严格的科学评估,确保上市产品的安全性和有效性,整个药品开发过程严谨、资料详尽。紧急状态下,研究者不可能从容地展开研究。更多的情况是研究者不得不在一个上市后的情境中去收集上市前的数据。以2009 年帕拉米韦的EUA 为例,FDA 安全性研究团队对EUA 期间采集的有关帕拉米韦的安全性数据进行了详尽分析。由于EUA 期间的临床应用并不是传统意义上的临床试验,其目的也不是采集临床数据,供安全性评估。EUA 期间,汇报至FDA 不良事件报告系统(ARES)中的不良事件是唯一的安全性数据。而且,临床和实验室检测资料也不完整。这些资料的不完整性,妨碍了FDA 判断不良事件是否与药物相关。FDA 研究人员的结论是EUA 期间的数据并不能满足安全性评估的需求,更多的信息须由正常的临床试验采集[12]。2010 年6 月,帕拉米韦的EUA 过期,帕拉米韦继续进行常规临床试验。2013 年帕拉米韦提交上市申请,2014 年最终在FDA 获得上市许可,彼时BioCryst 已于2012 年底前完成了2 项Ⅲ期临床试验。
显然,紧急状态下MCM 的安全性和有效性评估仍然面临很多挑战。随着科技和法规科学的进步,FDA 也在积极支持和尝试各种创新,例如建立哨兵首创项目(The Sentinel Initiative)、国家健康技术评估系统(The National Evaluation System for Health Technology)和移动交互式设备实时申请平台(The Real-Time Application for Portable Interactive Devices Platform)。这些基于电子健康数据和真实世界证据的基础设施的建立,可能在未来为评估MCM 提供机会[14]。
5 医疗应对产品供应保障
兵马未动,粮草先行。只有保障MCM 的充足供应,才能有力抗击公共卫生威胁。另一方面,稳定正常的医疗产品市场也不容懈怠。因此,我们有必要了解一下FDA 如何来保障医疗产品供应。
供应链监控:FDA 对于药品供应链一直都进行着积极监控。2019 年10 月,FDA 的CDER 在美国国会的证词对MCM 的供应链作了详细说明。FDA保有一个用于MCM 的药品清单,用于应对生物、化学、流感和放射性威胁。其中大部分药品都有战略储备。为保证在紧急状态下的充足供应,FDA 会采取各种措施,例如对MCM 原料药供应商进行上市后cGMP 检查,确保这些厂商的生产资质,及时发现并解决可能的问题。在公共卫生紧急状态时,FDA 也会积极寻求工业界合作,确保医疗应急品的生产和供应。FDA 将密切监控供应链情况,主动联系医药和器械生产厂,确保医疗产品供应链的安全,防止供应链的断裂或产品短缺。FDA 也将和各国和国际监管机构保持信息通畅,及早了解任何影响供应链的新情况的出现。如果医疗产品出现潜在短缺或供应断裂迹象,FDA 将会采取所有可能的措施,迅速反应,减少上述情况对病人以及医护人员的影响。例如,FDA 会和生产商密切合作增加产量,或加快评审可替代的供应商产品。
药品有效期延长计划:该措施使得美国政府机构,如国防部、SNS 等机构的储备物资在稳定性和质量得以保证的前提下,延长药品的保质期。在允许延长药品使用期限前,FDA 实验室人员将检测这些药品质量。在2018 财政年度,FDA 给予了大约2 100 个批次MCM 的保质期延长。
战略储备:FDA 也和SNS 协同为公卫紧急事件提供迅速充足的医疗物资供应。储备物资中未获批的研究用药或上市药的未获批适应证的使用,需要FDA 的EUA。2009 年甲型H1N1 流感大流行期间,SNS 完成了史上最大规模的物资部署,在全美范围内分发了大约1 250 万个疗程的抗病毒药品(另外还有30 万个疗程的抗病毒药品用于美国以外地区),1 950 万套个人防护设备,8 500 万只N95 口罩,2 129 个疗程的帕拉米韦针剂。SNS 中的储备物资都预先包装,一旦联邦部署令下达,所需物资可以在12 h 以内到达美国境内任何地方[15]。
先进生产技术:除了关注现有MCM 产能和储备,FDA 也很关注新兴、尖端生产技术在MCM 产能保障中的作用。FDA 认识到以连续性生产为代表的先进生产技术可以起到以下作用:①迅速提升疫苗和其他MCM 的产能;②缩短供应链,提高制备能力的弹性;③加速产品开发;④迅速提高包括细胞和基因疗法在内的新兴疗法的供应;⑤为应对药品短缺提供新的工具。我们以流感疫苗为例了解先进生产技术的优势。流感病毒毒株多、变异快,每年季节流行时都有数种毒株同时存在,而年年又不同。因此,流感疫苗需当年选取其中的3 ~ 4 个毒株进行设计制备。通常流感疫苗的制备需要数月时间,因此毒株的选取需要更早。先进生产技术在流感疫苗的生产中有以下优势:①使疫苗的制备更加接近流感季节,设计更有针对性的疫苗;②如果出现没有预测到的变化时,及时更换毒株;③在大流行中更快地生产;④疫苗短缺时可以容易地提高产能。此外,对于3D 打印这类尖端生产技术,FDA 也十分关注。这些先进技术的使用都可能在短时间内大幅提高生产商的制备能力,从而保障供应。
6 FDA 对公共卫生紧急事件的防范和反应
严重公共卫生事件,无论是传染性疾病的爆发、自然灾害的发生还是其他高危威胁,其发生发展都是一个快速变化的过程。FDA 作为医疗产品的监管机构,同时也监管着MCM,在应对公共卫生紧急事件中担负重要责任。该机构通过以下方式,在短时间内作出快速有效反应,并做好长远的防范和准备。
6.1 制定紧急行动计划
为有效应对可能出现的紧急情况,FDA 自2014年起开始制定《FDA 紧急行动计划》(FDA’s Emergency Operations Plan)[16]。该计划书详细地规定了紧急状态下如何在FDA 内部和其他部门间协调信息和资源。计划书每年更新,力求行动计划可以准确并符合当前需求,保证FDA 整个机构在紧急状态中可以高效运作,有效应对各种威胁。
6.2 法律保障下的快速反应
医疗产品是受到高度监管的产品,每一种产品的上市,必须经由FDA 全面科学评估,确保其安全性和有效性。但是公共卫生紧急事件出现时,现有的医疗产品一般不能满足需要。而公众又急需合适产品来应对紧急情况。为紧急应对公共健康威胁,公共卫生紧急状态宣布、EUA 等法律手段,允许FDA 将可能的MCM,包括在研产品或未获批准用途的产品,快速推向公众,为积极应对紧急事件提供必要手段。以此次新型冠状病毒肺炎(COVID-19)疫情为例,从美国发现第1 例患者起1 个月内FDA与相关部门的应对措施见表5。
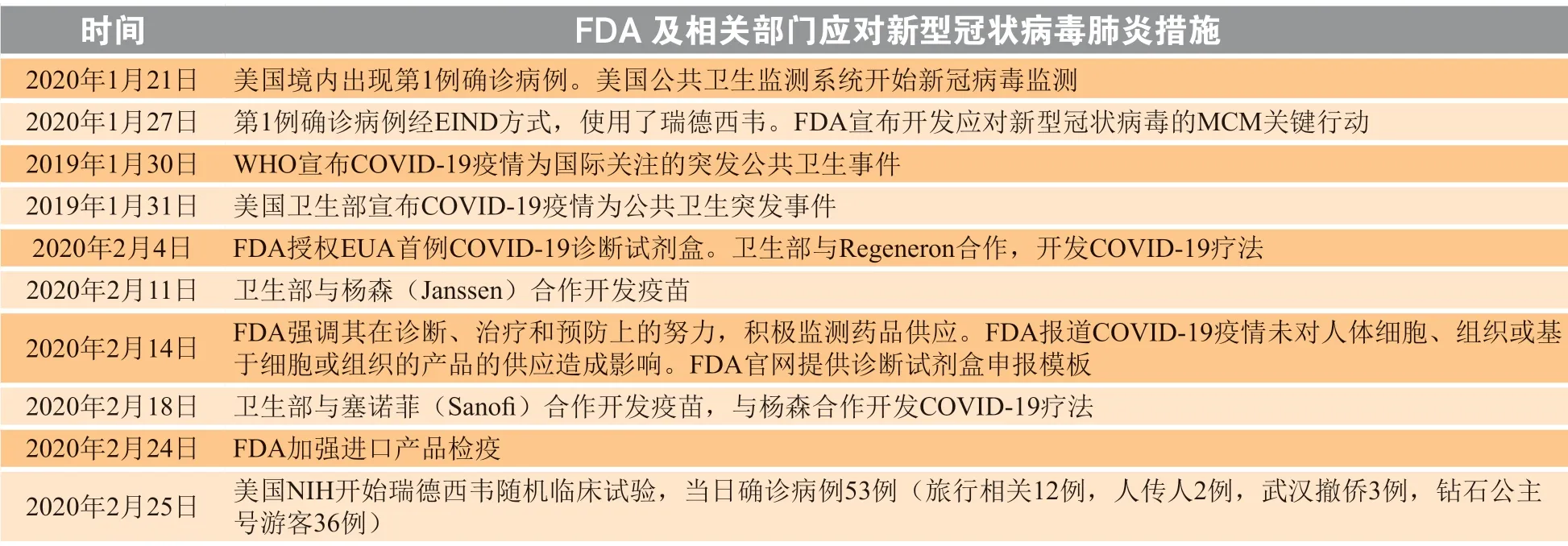
表 5 FDA 及相关部门应对新型冠状病毒肺炎的措施Table 5 Countermeasures against COVID-19 taken by FDA and related government partners
6.3 积极寻求医疗产品工业界支持
在重大公共卫生紧急事件中,美国政府会和工业界积极合作,确保MCM 的生产和供应。FDA 将密切监控供应链情况,了解生产商产能或短期大量增产可能,确保正常医疗产品供应,以及MCM 的供应。另一方面,FDA 也和工业界建立合作伙伴关系,加快MCM 的开发,帮助加快用于诊断、治疗和预防该疾病所需的医疗产品的问世,例如,在应对COVID-19 中与赛诺菲、杨森的紧密合作。而此前在应对埃博拉出血热、SARS、MERS 等重大流行性疾病时,默克、葛兰素史克、杨森、赛诺菲等大型药企都曾积极参与。以公共卫生紧急MCM 事业部(PHEMCE)为代表的美国政府部门十分了解与其他机构的合作。PHEMCE 早在2010 年的总结中就提到,政府、科研机构和药企更好、更早、更持久的合作可以有效扩大MCM 管线[17]。
6.4 鼓励研发的法规政策
疫情当中,FDA 采用EUA 的方式将MCM 快速推上“前线”。在疫情之外,FDA 更关注的是促进安全有效的MCM 的问世。FDA 会同卫生部下属PHEMCE,于2010 年开启了MCMi。这是FDA 内跨审评中心的一个项目,目的在于加速开发和使用安全有效的MCM,建立公共卫生和安全反应团队,发展MCM 法规科学,制定MCM 法律、法规和政策框架以及开展人员培训[3]。2015 年西非埃博拉疫情期间,FDA 通过MCMi 支持了相关医疗产品的评审和法规科学的研究。2017 年该项目支持了H7N9禽流感的防范措施。
6.5 政府资金支持
PHEMCE 隶属于美国卫生部,是一个跨部委部门,监管MCM 的科研、开发、采购、储备,制定有效使用MCM 的计划。PHEMCE 协调4 个部门[NIH、生物医学高级研发部(Biomedical Advanced Research and Development Authority)、SNS 和FDA]内MCM 工作。在2017—2021 年间,该部门的预算高达240 亿美元,主要公关方向有大流行性感冒(43 亿美元)、丝状病毒感染(包括埃博拉病毒)(16 亿美元)、天花病毒感染(12 亿美元)、化学威胁(12 亿美元)、放射和核威胁(18 亿美元)、广谱抗微生物剂(35 亿美元)、肉毒杆菌(3.8 亿美元)、炭疽(20 亿美元),以及其他项目(如寨卡病毒、中东呼吸综合征冠状病毒等)(26 亿美元)[18]。在这些公共资金的支持下,部分科研产品已成功转化为MCM。自2007 年, FDA 批准38 个产品用于应对生物、化学、放射性/核威胁和大规模流行性感冒[18]。而帕拉米韦在2009 年甲型流感大流行过去以后至2014 年获得FDA 批准上市前获得了美国政府超过5 亿美元的资助。最近,COVID-19 疫情爆发以来,卫生部下属生物医学高级研发部除了寻求和制药企业开发疫苗的合作,也在积极寻找开发诊断试剂的合作者,成功的合作者将得到75 万美元的政府资助。
6.6 长期备“战”
2017 年至今,美国卫生部一共宣布了48 次公共卫生紧急事件,其中新发事件32 次,16 次延长紧急状态[19],每次紧急事件规模和原因不尽相同。这些大大小小的紧急事件操练了从地方到联邦各级政府部门,使得各个部门可以协同工作,高效调动整合各种资源。以FDA 为例,为应对这些紧急事件,FDA 自2012 年批准了121 个MCM,而自2005 年颁布了超过60 个EUA。长期保持“战时”状态,充分锻炼了应急系统,积累了大量经验,人员得到锻炼,战备物资储备充分。一旦大疫情来临时,整个系统可以有序作出反应。
7 结语
2020 年的新冠肺炎疫情对于我国和全世界都是一次重大挑战。纵观最近100 年的世界历史,以流行性传染病为主要内容的公共卫生挑战越来越高发。虽然我们可以通过采取禁止食用野生动物的方式来减少动物源性疾病的发生,但我们不是生活在孤岛上,更多来源的传染性疾病依然威胁着人类,特别是对于我国沿海人口密集的大中型城市,一旦有流行性传染病的出现,将会迅速波及很大的人群。在此次抗击新冠肺炎疫情的过程中不仅存在着早期预警措施滞后,也存在着各种抗疫器材和物品如口罩、防护衣、酒精等产品供应不及时的问题。我国有着人口众多、人口居住集中和城际公共交通高效的特征,一旦出现呼吸道传染病,很容易扩展到更大的人群和其他城市。为此,建立高效的公共卫生应急措施和MCM 协调机制需要国家出面指导和参与储备。
它山之石可以攻玉。美国FDA 是一个多功能的机构,其管理着美国25%的市售产品。按照我国现行的行政体制,出现一个类似的强大行政机构并不容易。但在出现重大公共卫生挑战时,建立一个可以临时协调各部门的机构是非常必要且有效的。
尽管当下全球都在经历疫情的考验,美国疫情发展也非常迅速,但是FDA 在应急条件下的MCM审评供应体系依然很值得我们借鉴。当今的世界是一个经济全球化、人口流动全球化的世界。在这个人类共同体当中,没有一个国家在重大公共卫生威胁(特别是重大流行性传染病)中可以独善其身。各国间互相协作配合、学习、积极应对,必将在这个没有硝烟的战场上取得胜利。
致谢:本文在撰写及编校过程中,得到汉佛莱医药顾问有限公司贾清然女士的协助,特此感谢!
