分子印迹电化学传感器检测水杨酸
2020-05-08张夏红林水东卢晓春李锦辉
董 雁,张夏红,林水东,卢晓春,李锦辉
(1.龙岩学院 化学与材料学院,福建 龙岩 364012;2.固体废弃物资源化利用福建省高校 工程研究中心,福建 龙岩 364012)
分子印迹技术(Molecular impinting technology)是一种使聚合物聚合后与模板分子的空间构型相匹配的“空穴”结构的技术。所形成的“空穴”对模板分子有高度识别性,不易受外界干扰,稳定性与重现性好,可用于低浓度样品的检测[1]。分子印迹技术最早出现于生物化学,源于抗体对某种分子具有专一性识别功能的一种分析技术。之后,经过科学家的不断探索和完善,逐渐形成了一门新兴技术,得到了人们的普遍认可[2]。近年来,分子印迹技术在传感器领域的发展更可谓是动力十足,已成为该领域最具发展潜力的先进技术之一[3-5]。将电化学聚合法与分子印迹法相结合制备印迹聚合物具有使用仪器简单[6]、成本低、易操作等特点,且电化学聚合法制备的印迹膜厚度可控、成膜均匀、重复性较好,较化学聚合有更多优势。但该方法也存在一些不足,如未经修饰的玻碳电极表面灵敏度不高等。通常通过引入功能性纳米材料(如石墨烯等)以改善此类问题[7]。
水杨酸(SA)是一种脂溶性有机酸,为阿司匹林以及很多止痛药的制备原料,并被广泛用于化妆品中。但由于高浓度的SA会对皮肤造成损害,我国对化妆品中SA含量有明确规定,因此检测其在化妆品中的含量就尤为重要。传统检测SA的方法有荧光分光光度法[8]、气相色谱-质谱联用法[9]、高效液相色谱法[10-11]等。此外,也有基于印迹聚合技术检测SA的研究,但采用电聚合法制备SA分子印迹膜,用于检测SA含量却鲜见报道[12-15]。本文以邻苯二胺和吡咯为复合功能单体,SA作为模板分子,通过循环伏安法(CV)在石墨烯修饰的玻碳电极表面制备高选择性及高灵敏度的SA分子印迹膜。优化了分子印迹电化学传感器的制备条件,对传感器性能进行了表征,并采用传感器实现了对SA的检测。
1 实验部分
1.1 试剂与仪器
水杨酸(SA)、邻苯二胺(o-PPD)、乙腈(MeCN)、磷酸二氢钠、磷酸氢二钠、铁氰化钾、氯化钾、乙酸、乙酰水杨酸、对羟基苯甲酸、苯甲酸、吡咯(Py)、苯甲醛(分析纯,麦克林公司);0.5 mg/mL氧化石墨烯分散液(GO,南京先丰纳米材料科技有限公司)。
玻碳电极(GCE)、CHI660C电化学工作站(上海辰华仪器公司);KQ-100DE超声波清洗仪(昆山超声仪器有限公司);PH-3CB/3CU型PH计(上海越平科学仪器有限公司);S-3400N扫描电子显微镜(日本日立公司)。
1.2 石墨烯修饰玻碳电极的制备(GO/GCE)
先将玻碳电极分别用0.5 μm和0.05 μm的Al2O3粉末对电极表面抛光处理,然后用超纯水清洗3次,采用循环伏安法使其在铁氰化钾探针溶液中出峰电位差为85 mV后,自然风干。取5 μL氧化石墨烯分散液涂覆于预处理过的玻碳电极表面,自然晾干后再取3 μL滴涂至玻碳电极表面,自然晾干即制成石墨烯修饰玻碳电极(GO/GCE)[16]。
1.3 SA印迹电极的制备(MIP/GO/GCE)
在41 mL pH 6.6的磷酸缓冲溶液(PBS)中加入1 mL 0.1 mol/L SA溶液、4 mL 0.1 mol/Lo-PPD和4 mL 0.1 mol/L的Py溶液,充分混匀后待用。将“1.2”处理好的玻碳电极、参比电极及辅助电极所组成三电极体系浸入上述聚合液中,用循环伏安法(CV)扫描15圈,扫描范围为-0.2~0.8 V,扫描速度为50 mV/s进行制备。聚合结束后,先用超纯水反复冲洗电极,再放入洗脱液中洗脱20 min除去模板分子,自然晾干,得到SA分子印迹传感器(MIP/GO/GCE)。非印迹分子传感器(NIP/GO/GCE)的制备除不加SA外,其它条件保持不变。
2 结果与讨论
2.1 分子印迹聚合膜的电化学行为
采用CV法分别对NIP/GO/GCE、MIP/GCE及MIP/GO/GCE的电化学行为进行考察(图1)。结果显示,对NIP/GO/GCE进行第1次扫描时,在0.065 V处出现氧化峰,第2圈扫描后,电流强度明显降低,并随着扫描圈数的增多,电流恢复到背景值(图1A),表明o-PPD和Py可在此电流范围内发生聚合反应,在玻碳电极表面形成不导电的致密聚合膜,致使o-PPD和Py未进一步氧化,抑制了伏安电流响应。图1B 为MIP/GCE的印迹聚合行为,可观察到未加石墨烯时,修饰电极聚合的氧化还原峰不明显。而图1C为MIP/GO/GCE电聚合过程的CV图,可观察到强氧化还原峰,表明石墨烯具有良好的导电性,修饰电极后能提供给反应分子较大的比表面积,使反应更容易进行。
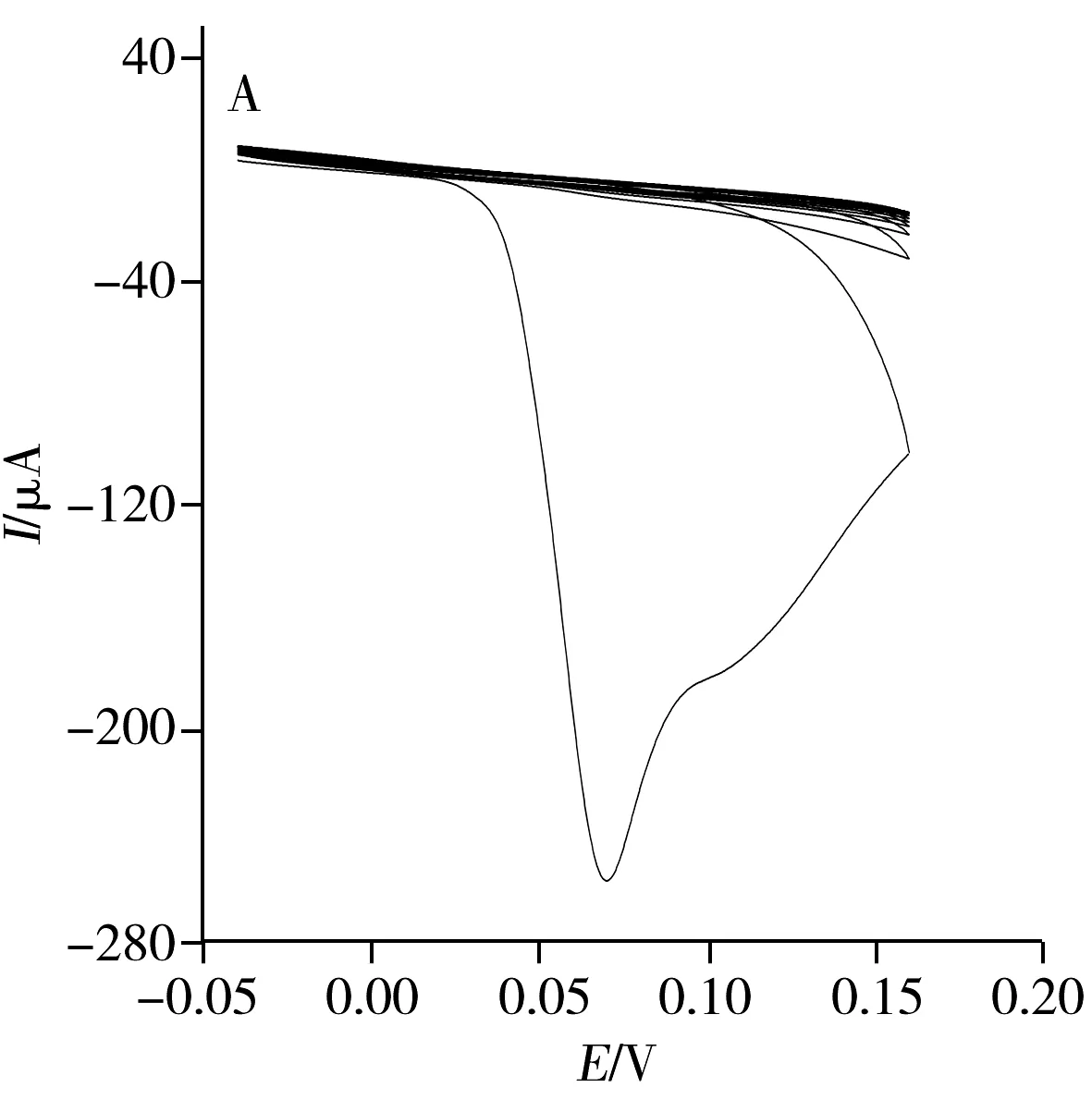

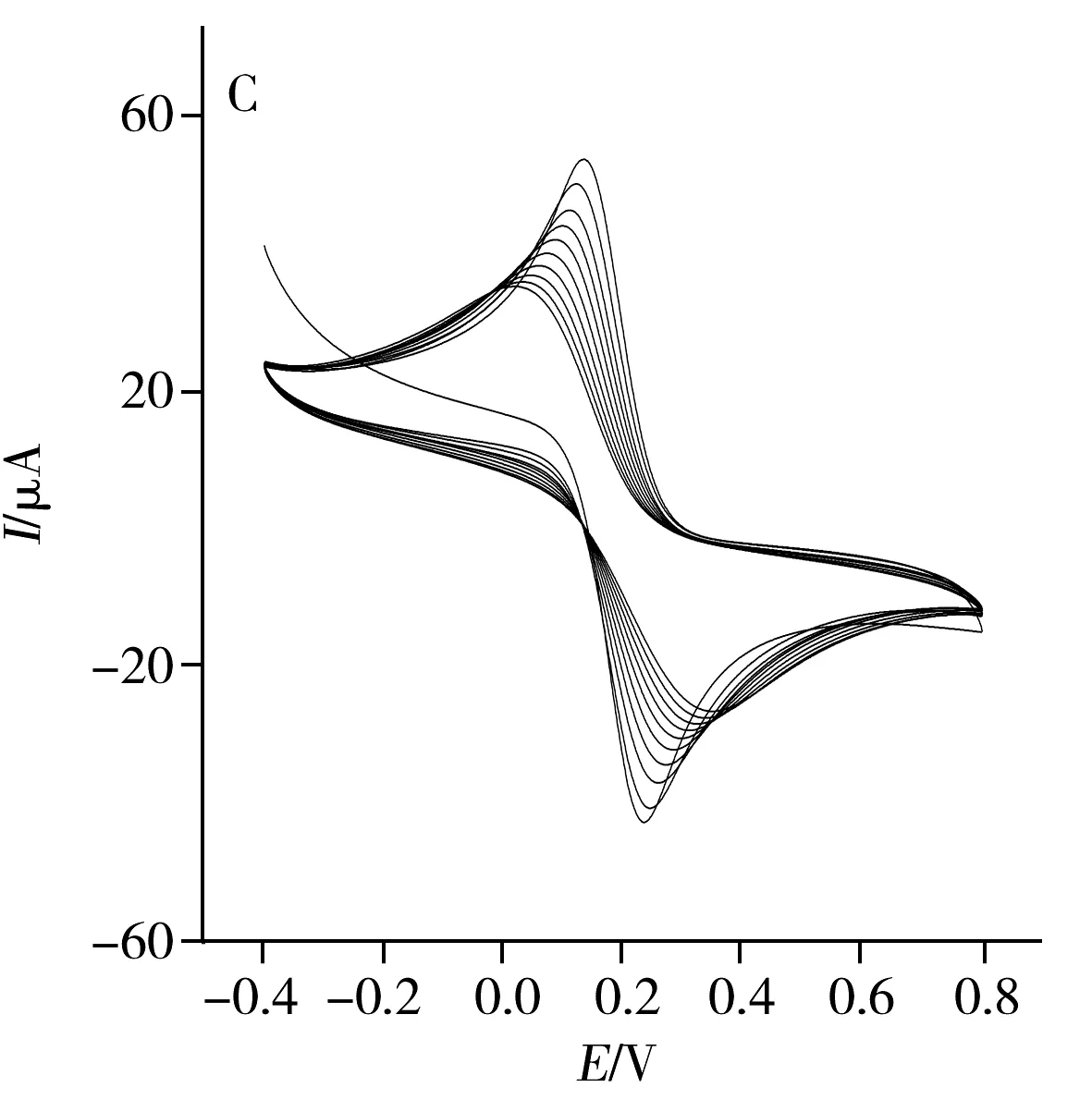
图1 NIP/GO/GCE(A)、MIP/GCE(B)及MIP/GO/GCE(C)的CV图Fig.1 CV curves of NIP/GO/GCE(A),MIP/GCE(B) and MIP/GO/GCE(C)
2.2 裸电极及石墨烯修饰电极的电化学表征

图2 不同类型电极在铁氰化钾探针溶液中的CV图Fig.2 CV curves of different electrodes in potassium ferrohydride probe solution a.GO/GCE;b,c.MIP/GO/GCE and NIP/GO/GCE after template removal,respectively;d,e.MIP/GO/GCE and NIP/GO/GCE before template removal,respectively
为了研究SA是否洗脱完全,考察了石墨烯修饰玻碳电极(GO/GCE)、洗脱前后的MIP/GO/GCE和洗脱前后的NIP/GO/GCE在K3[Fe(CN)6]探针溶液中的循环伏安图(如图2)。结果显示,GO/GCE的峰电流响应值最大(曲线a),未洗脱的电极MIP/GO/GCE(曲线d)和NIP/GO/GCE(曲线e)几乎无氧化还原峰,说明此电极基本绝缘,这是因为未洗脱的电极不会留下“空穴”,无法发生电子传递。洗脱后的电极MIP/GO/GCE,其峰电流响应显著增大(曲线b),这是因为印迹分子经洗脱剂洗脱后,在印迹膜上形成与模板分子匹配的“空穴”,形成电子传递的通道,因而导致峰电流信号明显加强。而洗脱后的MIP/GO/GCE表面覆盖膜虽具有“空穴”,但较GO/GCE的传递性能差,所以其电流响应的强度小于GO/GCE,但大于洗脱后的NIP/GO/GCE(曲线c)[17]。
2.3 不同类型印迹电极的扫描电镜图
采用扫描电镜分别对GO/GCE(A)、未洗脱的MIP/GO/GCE(B)、洗脱后的MIP/GO/GCE(C)和吸附SA平衡的MIP/GO/GCE(D)进行表征,结果如图3所示。从图A可以看出石墨烯的鳞片状结构;由图B可观察到聚合膜已将石墨烯的鳞片状结构覆盖,其表面比未聚合前平整;洗脱后的电极表面可观察到大量“空穴”(图C);吸附SA达到平衡后的MIP/GO/GCE电极,可观察到“空穴”数量变少,表面平整(图D),这是因为SA在静电力作用下与活性“空穴”结合[18]。此结论与“2.2”相互印证,说明石墨烯修饰后的玻碳电极的电流响应大于其他电极,未洗脱的电极表面则覆盖了一层不能导电的聚合物薄膜。
2.4 电化学聚合条件的选择
2.4.1 聚合液pH值的优化在浓度为0.2 mol/L的PBS缓冲溶液中,固定SA∶o-PPD∶Py的摩尔比为1∶4∶4,用方波伏安法研究了不同pH值(6.2、6.4、6.6、6.8、7.0)对聚合的影响。结果显示,在考察的pH值范围内,峰电流表现为先增大后减小的趋势,且在pH 6.6时峰电流响应值最大。说明在此pH值下,功能单体o-PPD和Py能较好的在修饰电极表面发生氧化聚合反应,其原因可能是o-PPD和Py更适宜在弱酸性环境下发生电化学聚合。 因此实验选择聚合液的pH值为6.6。


2.4.2 扫描圈数的优化扫描圈数直接关系修饰电极的聚合情况,通过改变扫描圈数可制备膜厚度不同的MIP/GO/GCE。在探针溶液中进行方波伏安法扫描,扫描圈数5~25,从峰电流变化情况考察电极聚合情况。结果显示,扫描5圈时印迹电极的峰电流响应值较小,这是由于扫描时间短,在玻碳电极表面形成的聚合膜较薄,当洗脱时能够得到的“空穴”较少[19],使得探针溶液中的[Fe(CN)6]3-能够结合的位点少,从而导致峰电流响应值较小。随着扫描圈数的增加,MIP/GO/GCE在探针溶液中的峰电流也增加,在扫描15圈时峰电流达到最大。继续增加扫描圈数,MIP/GO/GCE的峰电流呈递减趋势。这是由于扫描时间过长时,聚合膜变厚,使得在洗脱时洗脱液不易破坏模板分子与功能单体间的氢键[20],从而导致洗脱效果不好,峰电流下降。因此选择扫描圈数15为制备MIP/GO/GCE的最佳圈数。

图4 模板分子与功能单体的摩尔比对聚合的影响Fig.4 Influence of different ratios of template molecule to function monomers on polymerization
2.4.3 邻苯二胺与吡咯的比例优化聚合液中的模板分子与功能单体的比例会影响聚合膜的结构和电化学性能,因此需考察聚合液中模板分子与功能单体的比例,以寻找最佳配比。固定SA与o-PPD的最佳摩尔比为1∶4,改变Py的用量,使得聚合液中SA∶o-PPD∶Py的摩尔比分别为1∶4∶1、1∶4∶2、1∶4∶3、1∶4∶4、1∶4∶5、1∶4∶6,考察其峰电流响应情况,结果如图4。当SA∶o-PPD∶Py比例为1∶4∶4时,MIP/GO/GCE峰电流的响应值最大,说明此比例下有效结合位点最多,形成的印迹膜最稳定。因此,实验选择SA∶o-PPD∶Py的最佳摩尔比为1∶4∶4。

图5 不同洗脱液种类的洗脱效果Fig.5 Effect of different types of eluents
2.4.4 洗脱液的选择考察了混合洗脱液与单一洗脱液的洗脱效果,结果如图5所示,采用乙腈-乙酸(1∶1,体积比)、乙醇-乙酸(1∶1)混合液洗脱SA后,MIP/GO/GCE的峰电流与裸电极的峰电流几乎无差别,说明洗脱较完全,出现此现象可能是由于乙腈和乙醇均为优良的有机溶剂,在乙酸配合下可将大部分模板分子洗脱下来[21-22],甚至会导致印迹效果被破坏。采用用水-乙酸混合液(1∶1)虽效果略差,但相对较为温和,不会对印迹膜造成破坏。单一洗脱液氢氧化钠、乙醇、甲醇、水及乙酸洗脱后,峰电流响应较小(见图5),说明洗脱不完全,效果不佳。而甲醇-乙酸混合溶液(1∶1)的峰电流响应明显小于水-乙酸(1∶1)混合溶液,综上,本实验选择水-乙酸(1∶1)混合液为最佳洗脱液。
2.4.5 洗脱时间的优化采用方波伏安法考察了不同洗脱时间对MIP/GO/GCE峰电流的影响。结果显示,峰电流随着洗脱时间的增加而增加,在20 min时达到平衡,故选择最佳洗脱时间为20 min。
2.5 SA分子印迹传感器的性能研究
2.5.1 吸附平衡时间的确定将MIP/GO/GCE放入1.0×10-6mol/L SA溶液,采用方波伏安法研究了该电极对SA的峰电流以及与吸附时间的关系。结果显示,随着吸附时间的增加,SA分子在MIP/GO/GCE上的峰电流响应值呈现递增趋势,当吸附21 min后,峰电流趋于平稳,表明吸附21 min时反应达到平衡。为了吸附更加充分,实验选择吸附时间为24 min。
2.5.2 线性范围与检出限以MIP/GO/GCE为工作电极,在浓度分别为1.0×10-8、1.0×10-7、1.0×10-6、1.0×10-5、1.0×10-4、1.0×10-3、1.0×10-2mol/L 的SA溶液中,用方波伏安法测定SA浓度与峰电流的关系。结果显示,在1.0×10-8~1.0×10-2mol/L浓度范围内,MIP/GO/GCE的峰电流(Y)与SA浓度的负对数(lgc)呈线性关系,线性方程为Y=6.087 5×10-6+9.201 1×10-6lgc(r=0.998 1),检出限(S/N=3)为8.6×10-9mol/L。

图6 SA及其结构类似物在印迹传感器上的响应Fig.6 Selective responses of the imprinted electrochemical sensor for SA and its structural similarities
2.5.3 方法的选择性选择与SA结构类似的化合物(乙酰水杨酸、苯甲酸、苯甲醛、对羟基苯甲酸)作为干扰物进行选择性实验。分别配制浓度为5.0×10-6mol/L的SA、乙酰水杨酸、苯甲酸、苯甲醛、对羟基苯甲酸溶液,将MIP/GO/GCE在上述溶液中浸泡吸附24 min,自然干燥后采用方波伏安法在探针溶液中扫描。结果如图6所示,SA的峰电流响应值最小,这是因为MIP/GO/GCE上的“空穴”被SA“填满”,能结合探针溶液中[Fe(CN)6]3-的活性位点非常少,故峰电流响应值小。取10 μL上述干扰物质溶液加入空白PBS溶液中,用方波伏安法扫描印迹电极。由于SA在PBS溶液中以阴离子形式存在,在通电时与正性的“空穴”结合形成导电性“网格”,因此其电流响应值最大,而干扰分子则因空间构型与“空穴”不匹配[23],所以峰电流响应值较小。上述研究说明所制备的MIP /GO/GCE仅对SA有较高的选择性。
2.6 方法的重现性与稳定性
将MIP/GO/GCE置于浓度为1.0×10-7mol/L的SA溶液中,检测其峰电流响应值,每个标准样品平行测定6次,测得其相对标准偏差(RSD)为3.2%,说明MIP/GO/GCE的重现性较好。将MIP/GO/GCE置于pH 6.6的PBS溶液中存放1周后再检测1.0×10-7mol/L的SA溶液,其峰电流响应值基本稳定在原来的93%左右,说明MIP/GO/GCE的稳定性较好。
2.7 混合样品的检测
在39 mL空白PBS溶液中加入10 mL 1.0×10-6mol/L对羟基苯甲酸干扰溶液,再加入1 mL已知浓度为1.0×10-6mol/L的SA溶液,平行测定MIP/GO/GCE 3次,测得其加标回收率分别为101%、106%、102%,相对标准偏差为3.8%,说明此传感器可以用于实际样品的检测。
3 结 论
本文以SA为模板分子,o-PPD与Py作为复合功能单体,在石墨烯修饰的玻碳电极上制备对SA具有特定识别功能的电化学传感器。研究显示,SA在1.0×10-8~1.0×10-2mol/L浓度范围内,分子印迹传感器的峰电流与SA浓度的负对数呈良好的呈线性关系,其线性方程为Y=6.087 5×10-6+9.201 1×10-6lgc(r=0.998 1),检出限为8.6×10-9mol/L。将所制备的传感器用于混合样品检测,其回收率为101%~106%,相对标准偏差为3.8%。该方法操作简便,灵敏度高,为SA的提取、分离和检测提供了新思路。
