超临界水热极速合成异质结构H4SiW12O40/Bi2WO6光催化剂及其脱氮性能
2020-05-06邢宸
邢 宸
(大庆油田有限责任公司油田建设设计研究院, 黑龙江 大庆 163002)
随着中国经济的快速崛起,经济发展与环境保护的矛盾日益凸显[1,2]。中国作为石油消费大国,对成品油,尤其是轻质油中氧、氮、硫含量的要求愈加严格[3-6]。近年来,随着科研工作者对光催化氧化技术研究的不断深入,其性能稳定可靠,可控性强、能耗低、效率高、无二次污染等优势被广大科研工作者重视[7,8]。光催化剂在污水中有机物降解以及油品脱氮、脱硫等方面的应用也随着科研工作者的共同努力而日趋成熟[9-11]。但由于铋系光催化剂晶体结构的形成大多是先形成二维纳米片,再由二维纳米片逐渐自组装成棒状、球状、花瓣状等三维结构的一个缓慢生长过程,这就使得传统铋系光催化剂液相合成方法制备周期在24 h甚至更长[12,13],近年来,随着微波、超声波等技术在光催化剂制备方面的应用日趋成熟,但其制备周期仍需6 h左右,这就导致光催化剂的制造成本居高不下,严重影响了光催化技术在工业化领域的推广应用[14-16]。对传统光催化剂进行改性的诸多方法中,通过固载不同带隙光催化剂来调整整体活性的方式是最具前途的方式之一,而传统的浸渍固载方式固载物质与催化剂本体仅是依靠物理吸附力固定在一起,这就造成制得的光催化剂随着使用频次增加催化活性迅速下降[17-20]。异质结构的生成不但可以牢固把被固载物质固载在催化剂本体上,延长催化剂的使用寿命,而且异质结构可以起到两种不同晶相过渡带的作用,能够有效转移电子,提高光催化剂的催化活性,而且异质结构有助于降低光催化剂本体的光激发门槛,使复合光催化剂在可见光范围即可被激发产生较好的催化活性[21-23]。而如何快速高效地获得异质结构收率高、晶形好、光催化效率高的光催化剂成了广大科研工作者的奋斗目标[24]。
本研究采用超临界水热合成方式极速合成一种H4SiW12O40/Bi2WO6复合光催化剂,该复合光催化剂为二维纳米片自组装成的三维球状结构,其中,H4SiW12O40与Bi2WO6不是简单的固载关系而是在超临界水热条件下生成一种新的晶相Bi2SiWO2,正是由于这种晶相的存在,使得H4SiW12O40牢固固载在Bi2WO6光催化剂本体上的同时,对光生载流子进行了有效疏导,提升了H4SiW12O40/Bi2WO6光催化剂的使用寿命和光催化活性。针对光催化剂制备周期与晶形发育的矛盾,将超临界水热技术与光催化剂模板导向合成技术有机结合,在获得良好晶形异质结构H4SiW12O40/Bi2WO6光催化剂的同时明显缩短了光催化剂的制备周期,从而显著降低了催化剂的制备成本,攻克了光催化剂工业化应用的主要矛盾, 所制备的H4SiW12O40/Bi2WO6光催化剂轻质油脱氮效率达97%以上。
1 实验部分
1.1 试剂与仪器
无水乙醇, 广州化学试剂厂; 钨酸钠, 宜兴市化学试剂厂; 吡啶, 西安化学试剂厂; 硝酸铋, 泰州化学试剂厂; 硅钨酸, 天津市科密欧化学试剂有限公司; 石油醚, 天津化学试剂厂。以上试剂均为分析纯。
采用岛津公司 XRD-7000 型 X 射线衍射仪(Cu 靶,Kα射线, 管电压 40 kV, 管电流 30 mA,扫描速率 10(°)/min, 10°-80°扫描)对催化剂的晶相进行考察; 采用贝士德公司3H-2000BET-A型比表面积和孔隙度分析仪测定样品的比表面积和孔径分布; 通过美国Delong-LVem透射电镜观测晶体构造;通过德国卡尔蔡司公司的热场发射 SIGMA 型扫描电子显微镜观测样品粒子的微观形貌;采用北京市世纪森朗公司 TFM型高压反应釜制备催化剂(设计压力40 MPa,温度500 ℃ 内有磁力搅拌装置); 在上海比朗仪器有限公司 BL-GHX-V 光化学反应仪上进行光催化脱氮反应(光源为 500 W 氙灯, 内置恒温装置, 底部装有磁力搅拌装置)。
1.2 催化剂的制备
用100 mL去离子水溶解0.5 mol Bi(NO3)3·5H2O,并将Bi(NO3)3溶液置于超声波反应器中使其溶解均匀;用100 mL去离子水溶解0.25 mol Na2WO4·2H2O,并将Na2WO4溶液置于超声波反应器中使其溶解均匀;用100 mL去离子水溶解0.25 mol H4SiW12O40,并将H4SiW12O40溶液置于超声波反应器中使其溶解均匀;将配制好的Na2WO4溶液和H4SiW12O40溶液在磁力搅拌器的辅助下混合并搅拌均匀;在磁力搅拌器的辅助下将配置好的Na2WO4和H4SiW12O40混合液逐滴加入Bi(NO3)3溶液中;将混配好前驱液采用增压泵打入超临界水反应釜中,将反应釜增压升温至380 ℃、24 MPa(表压),然后迅速通过缓冲罐和节流阀节流降温即可得到干燥好的异质结构轻质油脱氮H4SiW12O40/Bi2WO6光催化剂。
1.3 催化剂的光催化脱氮性能测试
向沸程为 90-120 ℃的石油醚中加入一定量吡啶, 配制吡啶质量分数为 15 mg/g 的含氮模拟油,通过考察光催化剂对含氮模拟油的脱氮效果来评价所合成光催化剂的脱氮性能[25]。具体操作为: 将一定量的光催化剂和转子加入装有一定量模拟油的石英反应管中, 反应管口用套有塑料薄膜的胶塞塞紧, 置于光催化反应仪中。光催化反应仪底部有磁力搅拌装置, 反应仪光源由 500 W 氙灯提供, 内部设有冷却恒温装置, 通过调节循环水的流量对光催化反应仪中的温度进行调节, 从而实现恒温。反应结束后, 离心, 取上层清液, 采用国家石油产品碱性氮含量测定法(SH-T01629)检测碱性氮含量。
WNB=(V1-V0)N×14×1000/m
(1)
式中,WNB为碱性氮含量(mg/g);V0为空白参比样品所消耗的高氯酸-冰乙酸标准溶液的体积(mL);V1为滴定样品所消耗的高氯酸-冰乙酸标准溶液的体积(mL);N为高氯酸-冰乙酸标准溶液的质量浓度(g/mL);m为样品质量(g)。
2 结果与讨论
2.1 H4SiW12O40的固载量对H4SiW12O40/Bi2WO6光催化剂晶相、晶貌的影响
图1为超临界水热合成法固载不同含量H4SiW12O40的H4SiW12O40/Bi2WO6催化剂XRD谱图。由图1可知,H4SiW12O40的固载并没有影响载体催化剂Bi2WO6的斜方晶相(JCPDS No.26-1044,a=0.5480 nm、b=0.5480 nm、c=1.1500 nm),当H4SiW12O40固载量不超过20%时,随着H4SiW12O40固载量的增加,Bi2SiWO2特征峰逐渐增强,Bi2WO6各个特征峰并没有受到影响,反而在一定程度上增强,呈良性发展态势,这也说明了在一定程度上提高钨酸铋前驱液酸性有利于钨酸铋光催化剂的合成,当H4SiW12O40固载量超过20%时,随着H4SiW12O40固载量继续增加, Bi2SiWO2特征峰增强不明显,杂质峰反而凸显严重,斜方晶相Bi2WO6各个特征峰峰形受到影响,当H4SiW12O40固载量达到30%时,杂质峰凸显,斜方晶相Bi2WO6各个特征峰受到严重影响,这也说明过酸环境下不利于钨酸铋晶形的成长,故从XRD表征可以推断出,H4SiW12O40最佳固载量为20%。这也和文后所制备的不同H4SiW12O40固载量下的H4SiW12O40/Bi2WO6光催化脱氮性能对比测试相吻合。
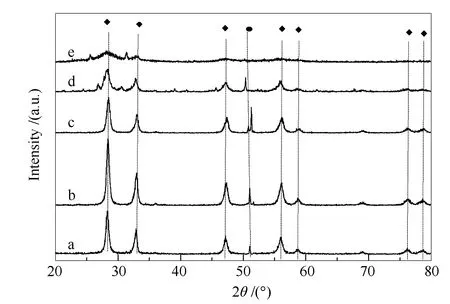
图1 采用超临界水热法制备的固载不同含量H4SiW12O40的H4SiW12O40/Bi2WO6光催化剂的XRD谱图
图2为最佳H4SiW12O40固载量下的H4SiW12O40/Bi2WO6的扫描电镜照片。由图2中可清晰地观察到颗粒之间相似度高,圆整度好,粒径为4.0-5.0 μm。在最佳H4SiW12O40固载量下的H4SiW12O40/Bi2WO6并没有出现较多的零散二维纳米片,大部分二维纳米片均已细密自组装成三维球状结构。这也和陈颖等[26]发现的在适当范围内对Bi2WO6前驱液酸度提高有利于其二维纳米片自组装成三维球状结构相一致。
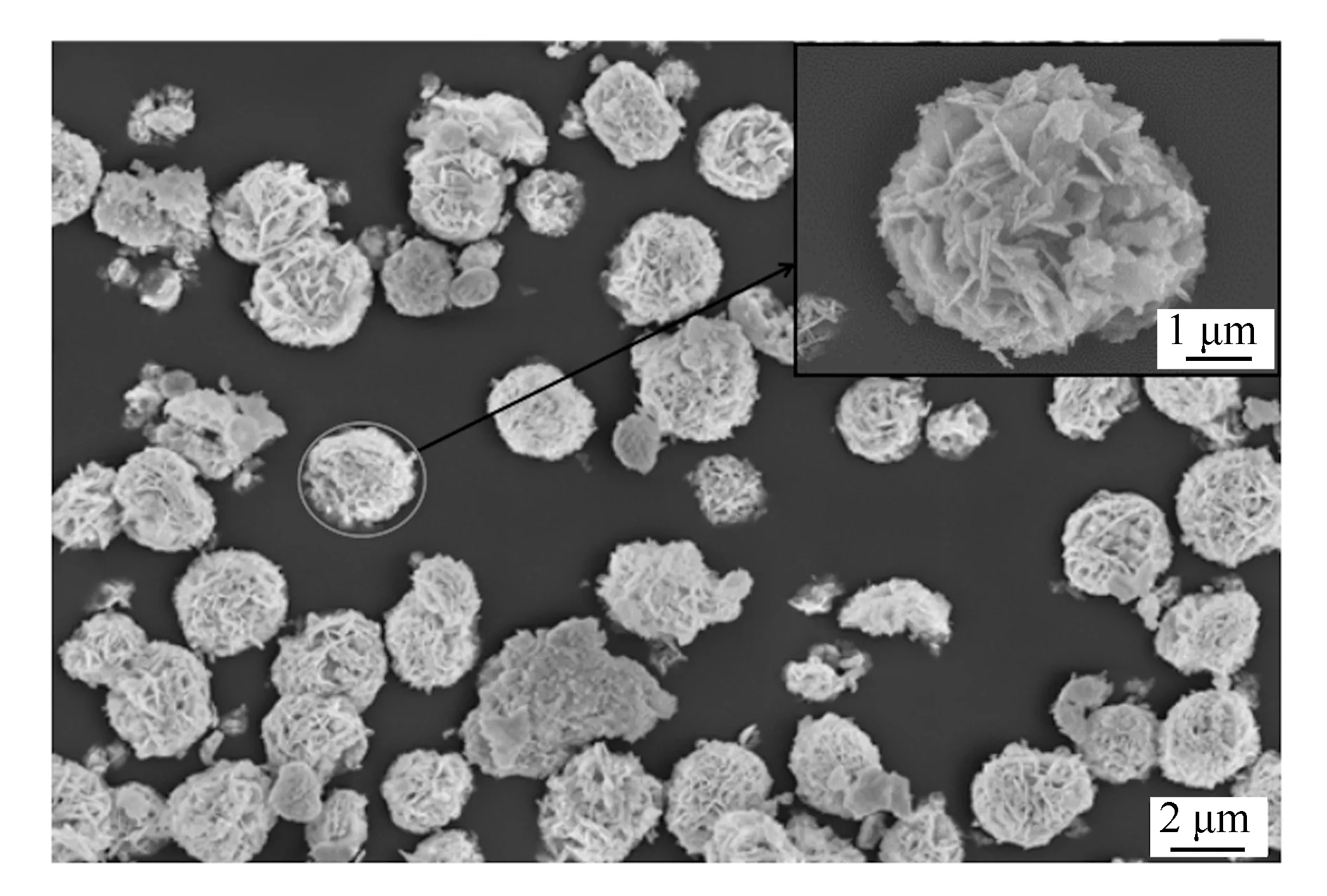
图2 采用超临界水热法制备的H4SiW12O40固载量(w=20%)的H4SiW12O40/Bi2WO6光催化剂的扫描电镜照片
图3为最佳H4SiW12O40固载量下的H4SiW12O40/Bi2WO6的透射电镜照片,由图3可以明显看出,所制备的H4SiW12O40/Bi2WO6光催化剂的三维球状结构是由二维纳米片组装堆叠而成,进一步观察可以看到二维纳米片为直径200 nm方形片状结构,局部出现的阴影部分晶面间距不同于Bi2WO6晶面间距和H4SiW12O40晶面间距,这也进一步印证了XRD表征Bi2SiWO2晶体生成的结果。
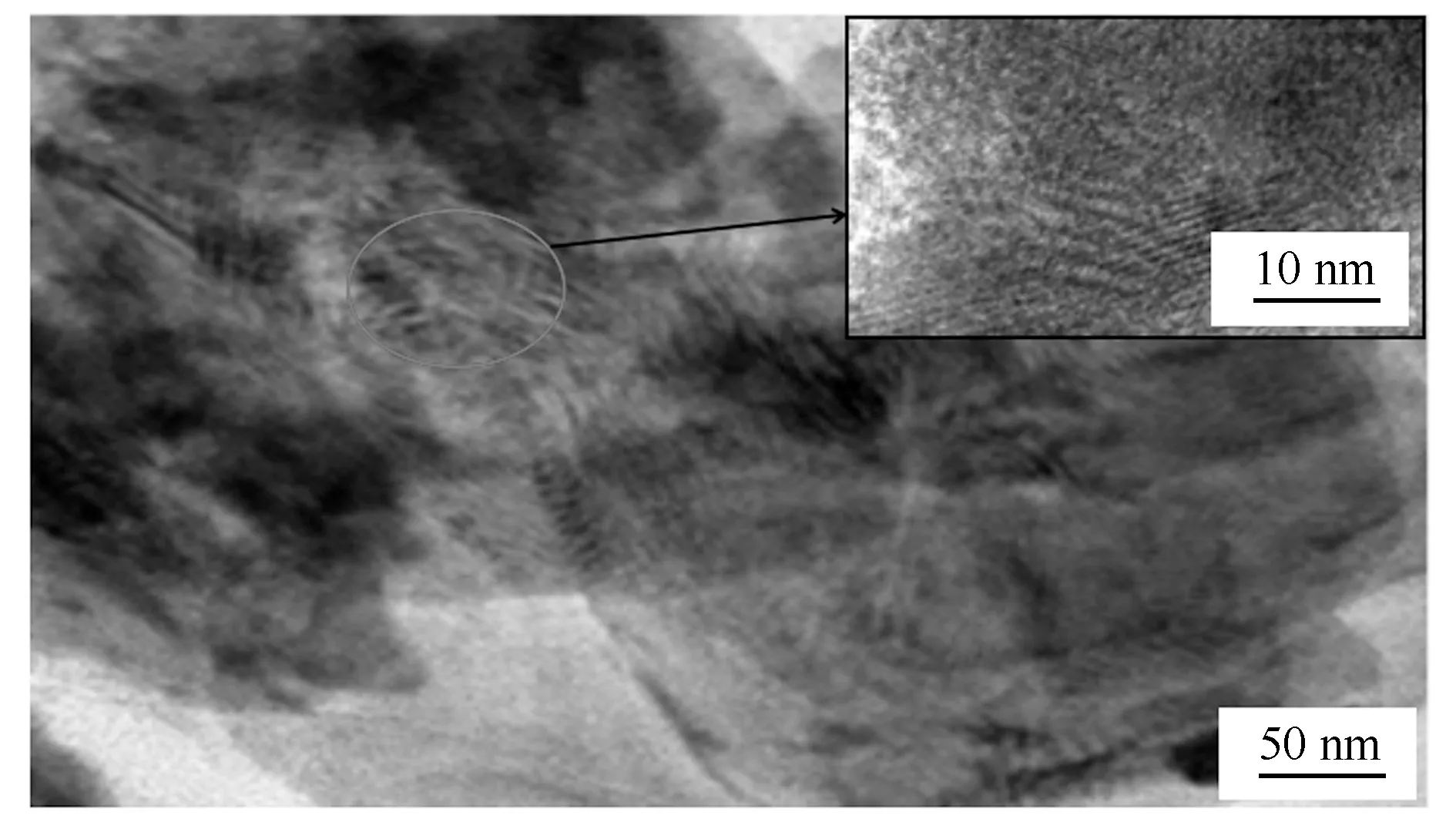
图3 采用超临界水热法制备的H4SiW12O40固载量(w=20%)的H4SiW12O40/Bi2WO6光催化剂的透射电镜照片
这种不同于H4SiW12O40晶体和 Bi2WO6晶体的新晶体的生成也进一步印证了之后H4SiW12O40/Bi2WO6光催化剂H4SiW12O40并非简单的物理吸附固载于Bi2WO6上,而是通过新物质的生成牢固地固载于Bi2WO6上,这种异质结构光催化剂相较传统浸渍法得到的物理吸附催化剂的光催化活性高且随着使用频次的增加催化活性基本没有衰减。
2.2 催化剂紫外可见漫反射表征及红移机理分析
图4三条紫外可见漫反射光谱谱图分别为,a:超临界水热法制备的最佳H4SiW12O40固载量下的H4SiW12O40/Bi2WO6光催化剂;b:传统水热浸渍法制备的H4SiW12O40/Bi2WO6光催化剂;c:水热法制备的Bi2WO6光催化剂。由图4可以观察到,相较Bi2WO6光催化剂单体,两种固载H4SiW12O40的光催化剂均出现不同程度的红移现象,这也证明固载H4SiW12O40后的光催化剂能被更低能量的光所激发,而相较传统水热浸渍法制备的H4SiW12O40/Bi2WO6光催化剂,超临界水热法制备的H4SiW12O40/Bi2WO6光催化剂红移现象明显,已经趋近500 nm,结合XRD表征进一步证明,H4SiW12O40和Bi2WO6本体并非简单的物理吸附关系而是通过这种不同于H4SiW12O40和Bi2WO6的Bi2SiWO2异质结构结合在一起,这种异质结构的生成有利于疏导光生载流子,并能使复合光催化剂互敏化作用增强,在可见光下即可被激发,降低光生载流子的光激发难度,提高光催化剂的光催化活性。H4SiW12O40和 Bi2WO6单体的禁带宽度分别为 3.00 和 2.74 eV,由式(2)算得H4SiW12O40/Bi2WO6的禁带宽度为 2.68 eV。

图4 不同方法制备的催化剂的紫外可见漫反射光谱谱图
αhv=A(hv-Eg)n/2
(2)
式中,α为吸附系数、h为普朗克常数、v为入射光频率、Eg为能隙、A为常量、n=1[27]。
H4SiW12O40在制备复合光催化剂的前驱液中提供适宜的酸性环境,起到良好的模板导向作用,并且H4SiW12O40是良好的电子载体与电子捕获剂,比轻质油中的氮化物更容易吸收光生电子从而变成激发状态,再将吸收的电子传递给氮化物从而变回基态[28]。这样硅钨酸就在氮化物和钨酸铋之间起到了良好的电子过渡作用, 能够利用自身一个基态-激发态-基态的循环, 迅速地将光生电子转移给氮化物而自身不损耗, 使得催化剂经光激发产生的电子和空穴得到了较好的分离,光生载流子复合效应明显下降,从而有效地提高了复合催化剂的光效率[29]; 而且H4SiW12O40本身也是一种具有一定光催化活性的物质, 经光激发后也可进行光催化反应,机理推断见图5。

图5 硅钨酸-钨酸铋耦合机理示意图
2.3 催化剂的比表面积及孔径分布
图6为超临界水热法制备的H4SiW12O40/Bi2WO6催化剂与传统液相合成法制备的H4SiW12O40/Bi2WO6催化剂氮气吸附-脱附等温线对比。由图6可知,在相对压力小于0.4时,氮气只是以单分子层形式吸附于催化剂表面,随着相对压力逐渐增加到0.4-0.9,发生多分子层吸附及毛细管凝聚吸附,图6中表现出明显的回滞环状,这也说明两种方式制备的催化剂均具有介孔结构,而采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂相较传统液相合成法制备的H4SiW12O40/Bi2WO6催化剂回滞环明显细窄,表明采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂介孔结构发育更加良好,大小均匀。
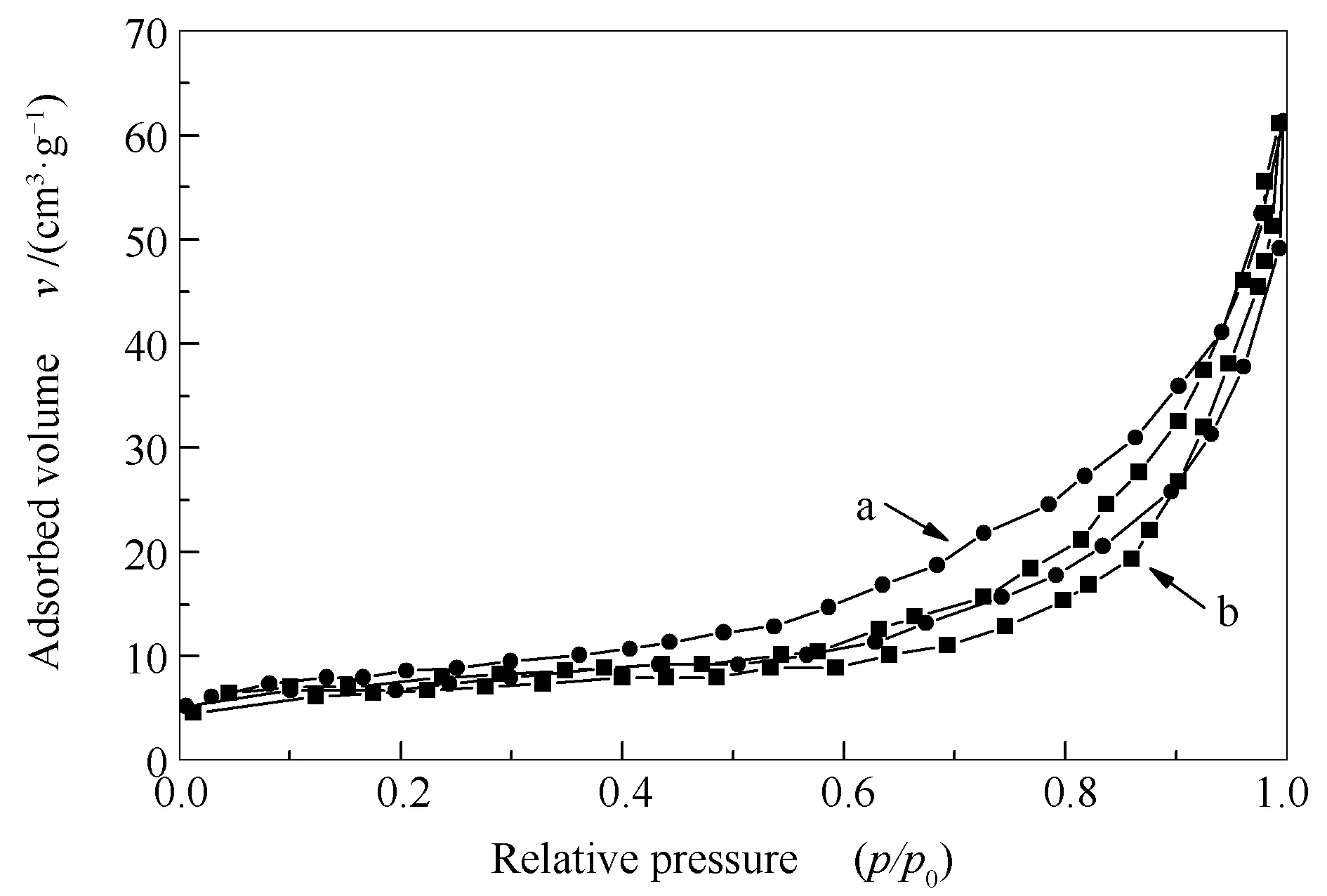
图6 传统水热法(a)和超临界水热法(b)制备的催化剂的氮气吸附-脱附等温线
图7为超临界水热法制备的H4SiW12O40/Bi2WO6催化剂与传统液相合成法制备的H4SiW12O40/Bi2WO6催化剂的孔径分布。由图7可知,相较传统液相合成法制备的H4SiW12O40/Bi2WO6,采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂孔径分布曲线较窄,这说明采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂孔径分布范围窄,进一步证明采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂孔径大小均匀的结论。催化剂的比表面积及平均孔径见表1。
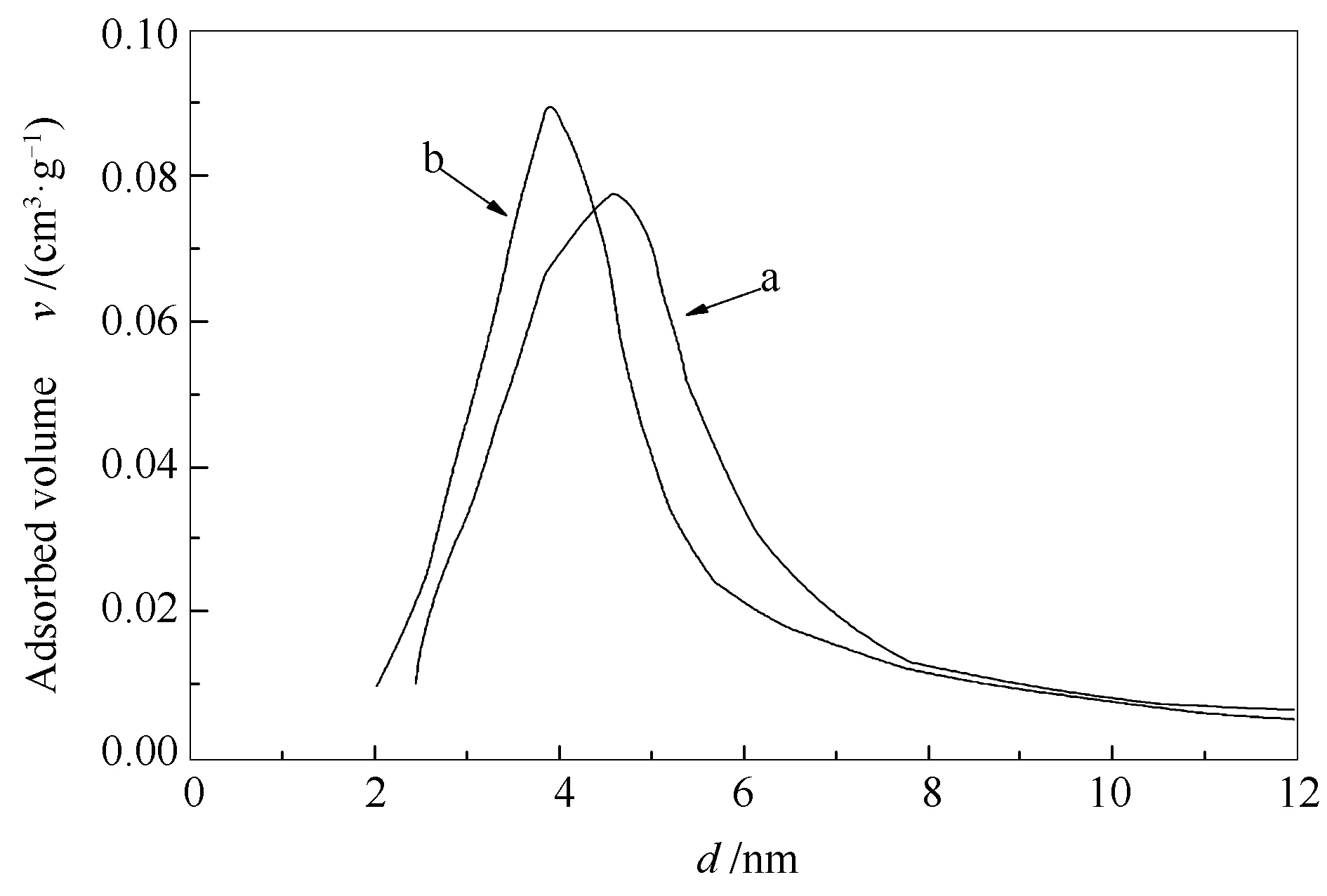
图7 传统水热法(a)和超临界水热法(b)制备的催化剂的孔径分布

表1 传统水热法(a)和超临界水热法(b)制备的催化剂比表面积及平均孔径
2.4 催化剂的脱氮性能
图8为超临界水热法制备的不同H4SiW12O40固载量H4SiW12O40/Bi2WO6光催化剂脱氮效率对比图。由图8可知,H4SiW12O40固载量不超过20%时,随着H4SiW12O40固载量增加,H4SiW12O40/Bi2WO6光催化脱氮性能逐渐增强,当H4SiW12O40固载量达到20%时, H4SiW12O40/Bi2WO6光催化脱氮性能达到峰值,对模拟油的脱氮效率达到97%,继续增加H4SiW12O40固载量,H4SiW12O40/Bi2WO6光催化脱氮性能下降明显,这也和固载不同含量H4SiW12O40的H4SiW12O40/Bi2WO6催化剂XRD谱图表征推测结论相一致。

图8 采用超临界水热制备条件下H4SiW12O40固载量对H4SiW12O40/Bi2WO6光催化剂脱氮性能的影响
图9为超临界水热法制备的最佳H4SiW12O40固载量H4SiW12O40/Bi2WO6催化剂与传统液相合成法制备的H4SiW12O40/Bi2WO6催化剂和Bi2WO6催化剂脱氮效率对比。由图9可知,固载H4SiW12O40的H4SiW12O40/Bi2WO6催化剂相较Bi2WO6催化剂脱氮效率明显提升,而采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂比传统液相合成法制备的H4SiW12O40/Bi2WO6催化剂脱氮性能好,这也和之前的表征结果一致。采用传统液相浸渍法制备的H4SiW12O40/Bi2WO6催化剂随着催化剂的使用频次增加催化效率明显衰减,而采用超临界水热法制备的H4SiW12O40/Bi2WO6催化剂随着催化剂使用频次的增加,催化效率基本没有变化,这也进一步验证了H4SiW12O40和Bi2WO6不是简单的物理吸附,而是通过分子间作用力结合在一起。
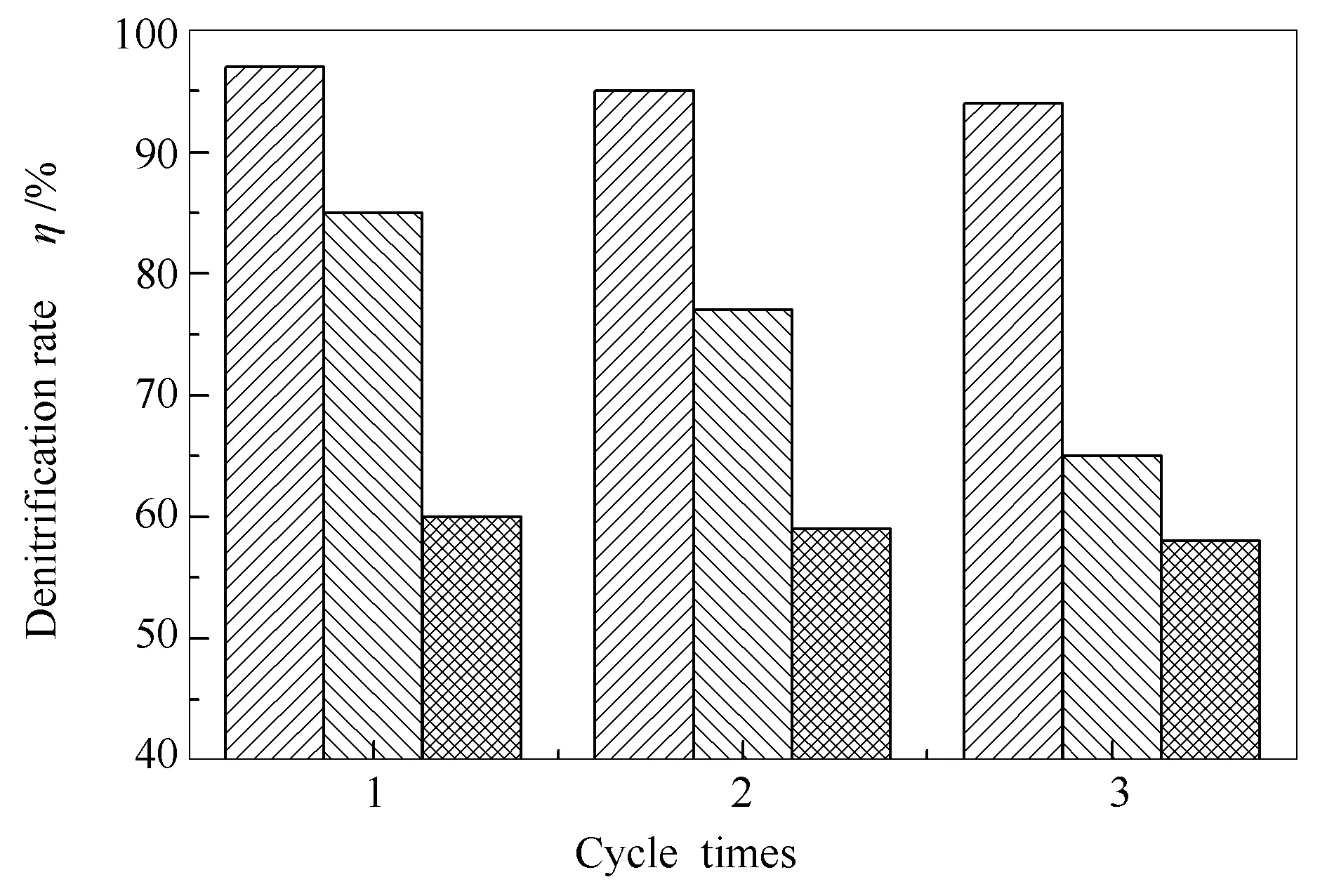
图9 不同方法制备的催化剂脱氮性能对比
2.5 催化剂制备周期对比
表2 为采用不同方法合成H4SiW12O40/Bi2WO6光催化剂制备周期对比。由表2可知,采用传统液相合成方式耗时较长,需要12 h左右,近两年,随着微波辅助液相合成方式的采用,虽然能够明显缩短催化剂制备周期,但仍然需要6 h左右,使得催化剂制备成本居高不下。本研究采用的超临界水热法利用水在超临界条件下的超高均相性、较高的分子迁移率、自催化性等,在10 s即可获得晶象好、比表面积大、空隙分布均匀的H4SiW12O40/Bi2WO6光催化剂,并且所制备的催化剂干燥清洁,无需进一步处理,明显缩减了催化剂的制备周期,从而降低催化剂的制备成本,推广了工业化应用。

表2 不同方法制备周期对比
3 结 论
本研究主要是针对光催化剂制备周期与晶形发育的矛盾,在超临界水环境下利用硅钨酸作为模板导向剂和固载原料合成具有异质结构的H4SiW12O40/Bi2WO6,在获得高脱氮活性H4SiW12O40/Bi2WO6光催化剂的同时把超临界水的独特理化性质与催化剂的合成工艺技术有机结合,在10 s内获得晶形发育良好的脱氮效率达97%以上的H4SiW12O40/Bi2WO6异质结构光催化剂,明显缩短了催化剂的制备时间,从而降低催化剂的制造成本,显著地推动了光催化剂由于制备周期长、晶形不好而造成的工业化应用率低的技术难题。
