淫羊藿根质量标准的研究
2020-04-29万红才徐作刚罗洪莲刘晓艳徐文芬
段 萍,万红才,徐作刚,罗洪莲,刘晓艳,徐文芬
(1.黔南州检验检测院,贵州 都匀 558000;2.贵州中医药大学,贵州 贵阳 550000)
淫羊藿Epimedium brevicornuMaxim 最早载于《神农本草经》[1],生于海拔200~1 800 m 的山坡、草丛、灌丛、沟谷、岩石缝、林下等,分布于贵州、四川、长江流域中下游及南方各省区,其根为应用历史悠久的传统中药,也是贵州常用的苗药,重在补虚、祛风湿,具有补肾阳、强筋骨、助阳益精等功效,主治肾虚阳痿、小淋沥、喘咳、风湿痹痛。淫羊藿含有多种黄酮类活性成分,主要是8-异戊烯基黄酮,为该属植物特有的黄酮类成分,包括双藿苷A、淫羊藿属苷A、朝藿定C、淫羊藿苷、淫羊藿属苷C 等,目前从巫山淫羊藿根、粗毛淫羊藿根、黔岭淫羊藿根也分离到该类化合物;淫羊藿总黄酮具有抑制破骨细胞、促进成骨细胞生长、抗抑郁、增强免疫调节、保护心脑血管系统、抑菌、抗炎、抗病毒、抗氧化、抗衰老、抗肿瘤等药理活性[2-4]。
2003 年版《贵州省中药材、民族药材质量标准》 收载了箭叶淫羊藿、巫山淫羊藿、粗毛淫羊藿、柔毛淫羊藿、天平山淫羊藿、毡毛淫羊藿、光叶淫羊藿的干燥根作为药用资源[5],但其质量标准中仅有性状描述。本实验对淫羊藿根来源、性状、鉴别、检查、含有量测定等方面进行研究,以期进一步完善其质量标准。
1 材料
1.1 仪器 TΜ-1901 紫外分光光度计(北京普析通用仪器有限责任公司);UV-2600 紫外分光光度计、2010 AHT 高效液相色谱仪(日本岛津公司);SK250LHC 超声波清洗器(上海科导仪器有限公司);卡玛TLC PLATE HEATER Ⅲ薄层板加热器;AG135 电子天平(十万分之一)、AL204 电子天平(万分之一)(瑞士梅特勒-托利多公司);安捷伦1260 高效液相色谱仪(美国安捷伦公司)。
1.2 试剂与药物 朝藿定C(纯度92.6%,批号111780-201803)、淫羊藿苷(纯度94.2%,批号110737-201516)对照品均购于中国食品药品检定研究院,2~10 ℃冷藏;硅胶G 板、硅胶GF254板、硅胶H 板均为青岛海洋化工有限公司生产。乙腈为色谱纯;稀乙醇为50% 乙醇;其他试剂为分析纯;水为娃哈哈纯净水。样品洗净后晒干,粉碎,过3 号筛,置于干燥器中保存,经贵阳中医学院何顺志教授、王悦云副教授鉴定为正品。
2 方法与结果
2.1 来源 本品为淫羊藿属中8 个品种的干燥根,夏、秋两季采挖,洗净,晒干。具体见表1。
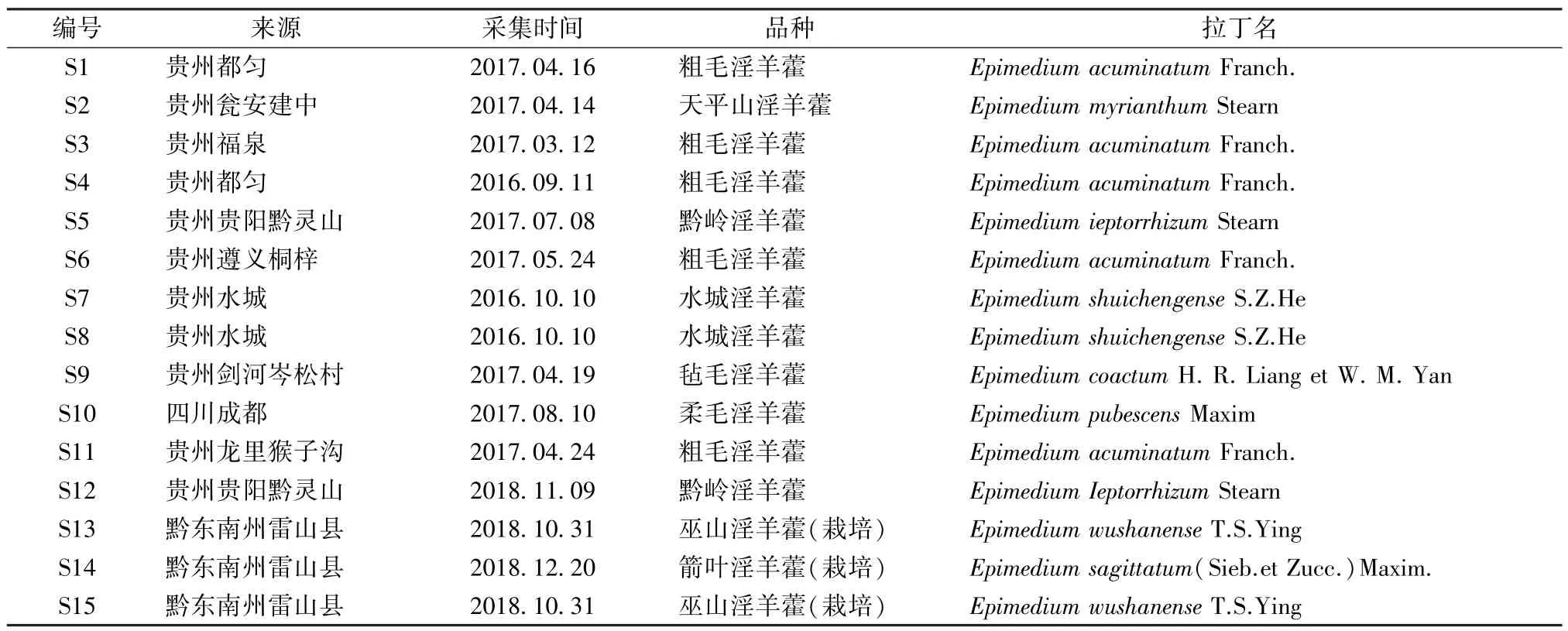
表1 样品信息Tab.1 Information of samples
2.2 性状 对8 个品种淫羊藿根的性状进行描述,结果见表2。
2.3 鉴别
2.3.1 显微鉴别 本品粉末棕黄色;导管成束或散在,直径10~50 μm,螺纹或网纹,导管分子穿孔板上具多数圆形孔;薄壁细胞类方形、类长方形、不规则形,壁稍厚,具纹孔,多含黄棕色分泌物;木栓细胞类长方形、类方形,直径10~50 μm,具纹孔;木纤维成束或单个散在,呈宽披针形。见图1。
2.3.2 薄层色谱 取本品细粉0.2 g,加乙醇10 mL,超声30 min,滤过,滤液蒸干,残渣加1 mL甲醇溶解,作为供试品溶液;另取朝藿定C对照品适量,加甲醇制成每1 mL 含1 mg 该成分的对照品溶液。按照2015 年版《中国药典》 四部通则0502 薄层色谱法,吸取上述2 种溶液各5 μL,点于同一硅胶G 薄层板上,以三氯甲烷-甲醇-水(27∶12∶4)下层澄清溶液为展开剂,展开,取出,晾干,喷以10%硫酸乙醇溶液,于105 ℃加热至斑点显色清晰。结果,供试品色谱在对照品相应位置上显相同颜色的斑点,见图2。
2.4 检查 按照2015 年版《中国药典》 四部通则,对水分(通则0832 第二法)、总灰分(通则2302)、酸不溶性灰分(通则2302)进行检查,结果见表3。由于淫羊藿圆柱型根分支少,易清洗;结节型根包裹杂质较多,不易清洗,导致总灰分、酸不溶性灰分测定结果偏差较大,暂定水分含有量不得超过12.0%,总灰分含有量不得超过9.0%,酸不溶性灰分含有量不得超过6.0%。

表2 淫羊藿根外观性状Tab.2 Appearance characteristics of the roots of E.brevicornu

图1 淫羊藿根显微鉴别Fig.1 Microscopic identification of the roots of E.brevicornu
2.5 浸出物 按照2015 年版《中国药典》 四部通则2201 项下醇溶性浸出物测定法中的热浸法测定,用稀乙醇作为溶剂,结果见表3,暂定浸出物含有量不得少于18.0%。

图2 淫羊藿根TLC 色谱图Fig.2 TLC chromatogram of the roots of E.brevicornu
2.6 含有量测定
2.6.1 总黄酮[6]
2.6.1.1 测定方法 精密量取朝藿定C 测定项下的供试品溶液0.5 mL,置于50 mL 量瓶中,加甲醇至刻度,摇匀,作为供试品溶液。另取淫羊藿苷对照品适量,精密称定,加甲醇制成每1 mL 含10 μg该成分的对照品溶液。取2 种溶液,以相应试剂为空白,按照紫外-可见分光光度法(通则0401)在270 nm 波长处测定吸光度,计算含有量。
2.6.1.2 对照品溶液制备 精密称取淫羊藿苷对照品10.43 mg,置于100 mL 量瓶中,甲醇超声溶解并稀释至刻度,作为贮备液,精密量取1 mL,置于10 mL 量瓶中,加稀乙醇稀释至刻度,即得(9.825 1 μg/mL)。
2.6.1.3 供试品溶液制备 精密量取朝藿定C 测定项下的供试品溶液0.5 mL,置于50 mL 量瓶中,加甲醇至刻度,摇匀,即得。
2.6.1.4 线性关系考察 精密量取“2.6.1.2”项下贮备液,甲醇制成1.965、4.913、9.825、19.650、29.475 μg/mL,按“2.6.1.1”项下方法测定吸光度。以溶液质量浓度为横坐标(X),吸光度为纵坐标(Y)进行回归,得回归方程为Y=0.041 5X+0.007 4(r=0.999 9),在1.965~29.475 μg/mL 范围内线性关系良好。
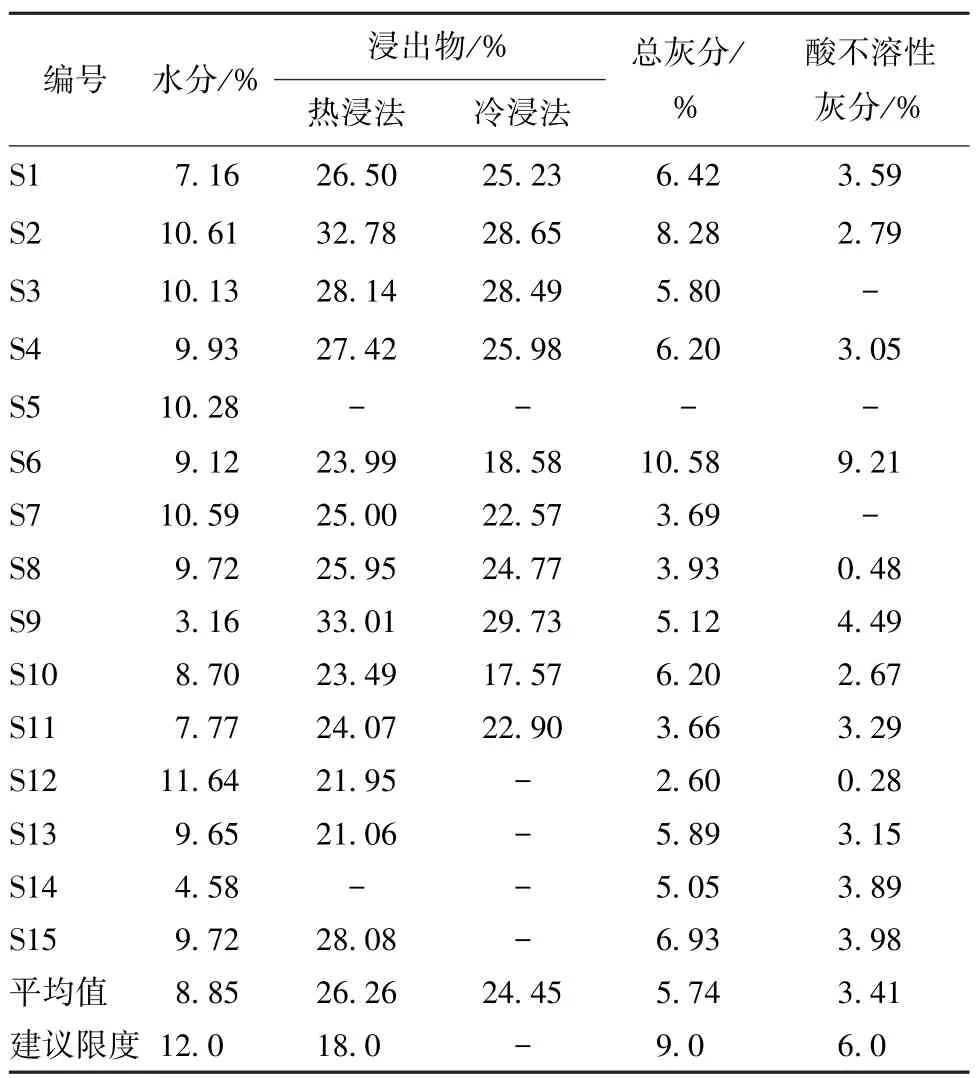
表3 水分、总灰分、酸不溶性灰分、浸出物测定结果Tab.3 Determination results of moisture,total ash,acid insoluble ash and extract
2.6.1.5 精密度试验 精密吸取“2.6.1.2”项下对照品溶液,按“2.6.1.1”项下方法测定吸光度6 次,测得其RSD 为0.01%,表明仪器精密度良好。
2.6.1.6 稳定性试验 取样品(S1),按“2.6.1.3”项下方法制备供试品溶液,按“2.6.1.1”项下方法于0、6、12、16、20 h 测定吸光度,测得其RSD为0.7%,表明溶液在20 h 内稳定性良好。
2.6.1.7 重复性试验 按“2.6.2.3”项下方法制备6 份供试品溶液,各取0.5 mL 置于50 mL量瓶中,按“2.6.1.3”项下方法制备供试品溶液,按“2.6.1.1”项下方法测定吸光度,测得淫羊藿苷含有量RSD 为1.5%,表明该方法重复性良好。
2.6.1.8 加样回收率试验 称取淫羊藿苷对照品78.22 mg,置于150 mL 量瓶中,加稀乙醇至刻度,作为回收对照品溶液(491.22 μg/mL)。取6 份样品(S1),每份约0.1 g,精密称定,置于具塞锥形瓶中,加入回收对照品溶液20 mL,按“2.6.1.3”项下方法制备供试品溶液,按“2.6.1.1”项下方法测定吸光度,计算回收率,结果见表4。
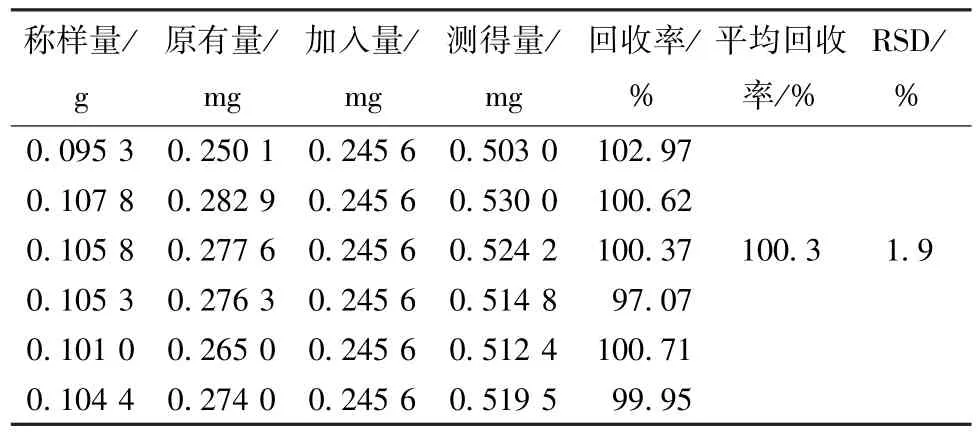
表4 总黄酮加样回收率试验结果Tab.4 Results of recovery tests for total flavonoids
2.6.1.9 样品含有量测定 取15 份供试品溶液,各量取0.5 mL,置于50 mL 量瓶中,按“2.6.1.1”项下方法测定吸光度,以淫羊藿苷为基准计算含有量(按干燥品计),结果见表5。参照2015 年版《中国药典》 一部淫羊藿总黄酮测定项下规定限度,建议将含有量限度暂定为按干燥品计算,含淫羊藿苷(C33H40O15)不得少于6.0%。
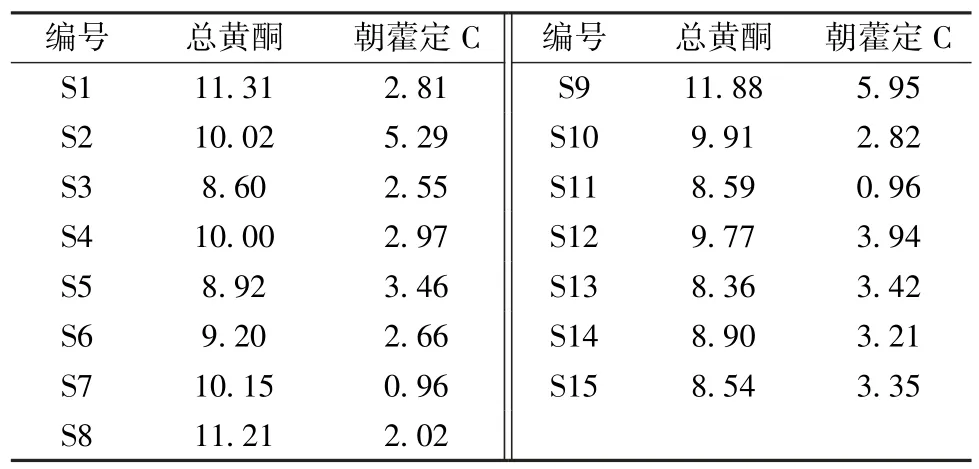
表5 各成分含有量测定结果(%)Tab.5 Results of content determination of various constituents(%)
2.6.2 朝藿定C[7-9]
2.6.2.1 色谱条件 十八烷基键合硅胶色谱柱(4.6 mm×250 mm,5 μm);流动相乙腈-水(25∶75);体积流量1.0 mL/min;柱温30 ℃;检测波长270 nm;进样量10 μL。理论塔板数按朝藿定C峰计,应不低于3 000。见图3~4。
2.6.2.2 对照品溶液制备 精密称取朝藿定C 对照品13.17 mg,置于25 mL 量瓶中,稀乙醇超声溶解并稀释至刻度,作为贮备液,量取5 mL,置于10 mL量瓶中,乙醇稀释至刻度,即得(0.244 mg/mL)。
2.6.2.3 供试品溶液制备 取样品(S1)约0.2 g,精密称定,置于具塞锥形瓶中,精密加入稀乙醇20 mL,称定质量,静置过夜,超声60 min,放冷,稀乙醇补足减失的质量,摇匀,0.45 μm 微孔滤膜过滤,取续滤液,即得。
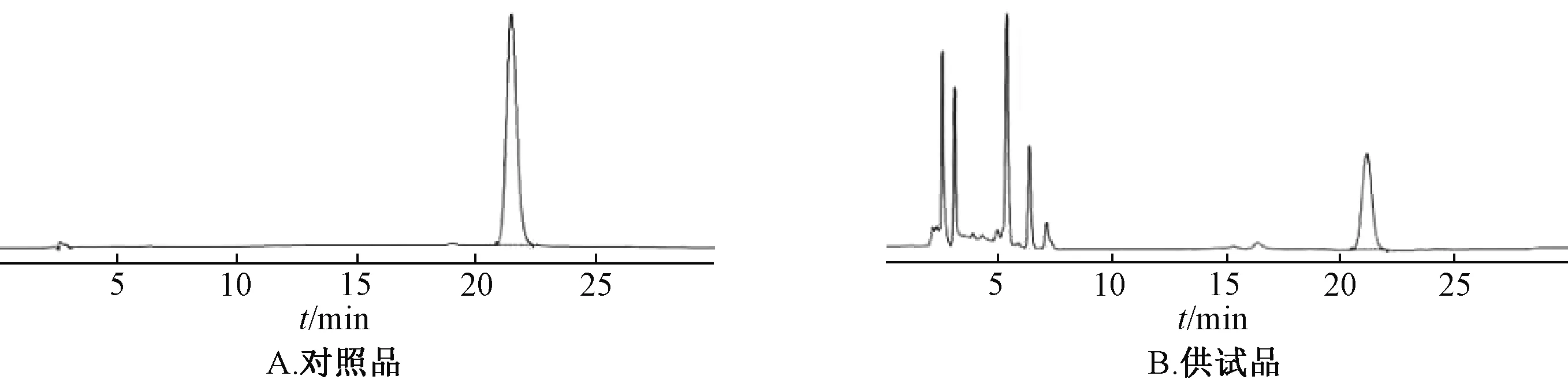
图3 淫羊藿根HPLC 色谱图Fig.3 HPLC chromatograms of the roots of E.brevicornu
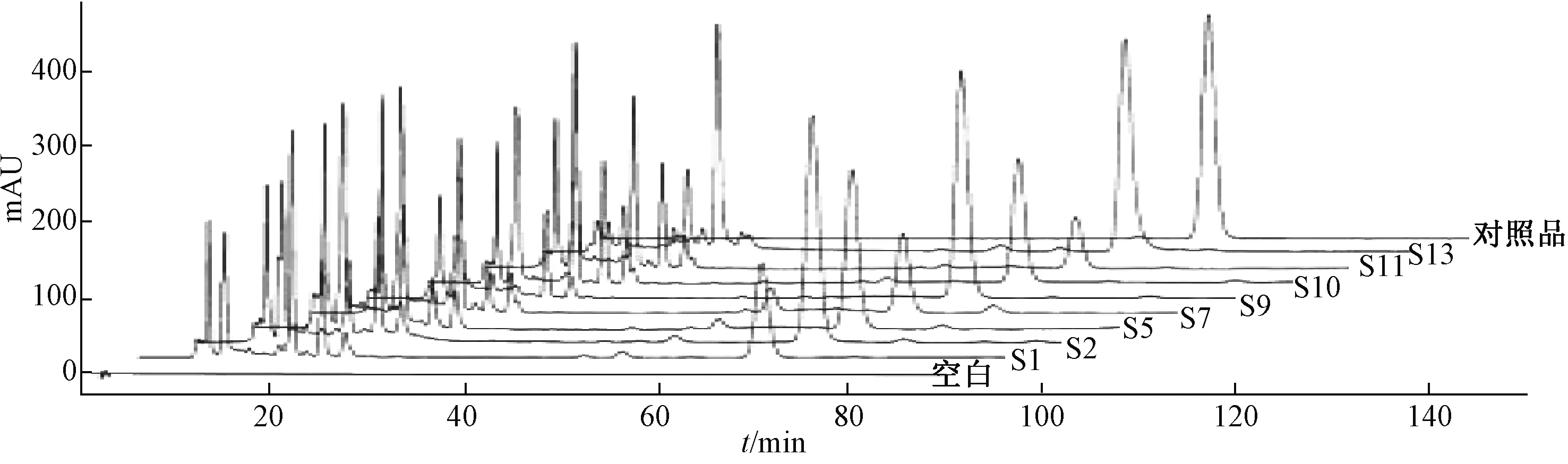
图4 各样品HPLC 色谱图Fig.4 HPLC chromatogram of various samples
2.6.2.4 线性关系考察 精密量取“2.6.2.2”项下贮备液 2、4、8、12、16、20 μL,在“2.6.2.1”项色谱条件下测定。以峰面积为纵坐标(Y),进样量为横坐标(X)进行回归,得方程为Y=1 766.284X-121.810(r=1.000 0),在0.976~9.756 μg 范围内线性关系良好。
2.6.2.5 精密度试验 精密吸取“2.6.2.2”项下对照品溶液,在“2.6.2.1”项色谱条件下连续进样6 次,测得朝藿定C 峰面积RSD 为0.18%,表明仪器精密度良好。
2.6.2.6 稳定性试验 取供试品溶液(S1),在“2.6.2.1”项色谱条件下于0、6、12、16、20 h进样,测得朝藿定C 峰面积RSD 为0.7%,表明供试品溶液在20 h 内稳定性良好。
2.6.2.7 重复性试验 取样品(S1)6 份,精密称定,按“2.6.2.3”项下方法制备供试品溶液,在“2.6.2.1”项色谱条件下测定,测得朝藿定C 含有量RSD 为1.4%,表明该方法重复性良好。
2.6.2.8 加样回收率试验 称取朝藿定C 对照品23.80 mg,置于200 mL 量瓶中,加稀乙醇适量,超声溶解,放冷,加稀乙醇至刻度,作为回收对照品溶液(0.110 2 mg/mL)。取6 份样品(S1),每份约0.1 g,精密称定,置于具塞锥形瓶中,加入回收对照品溶液20 mL,按“2.6.2.3”项下方法制备供试品溶液,在“2.6.2.1”项色谱条件下测定,计算回收率。结果见表6。
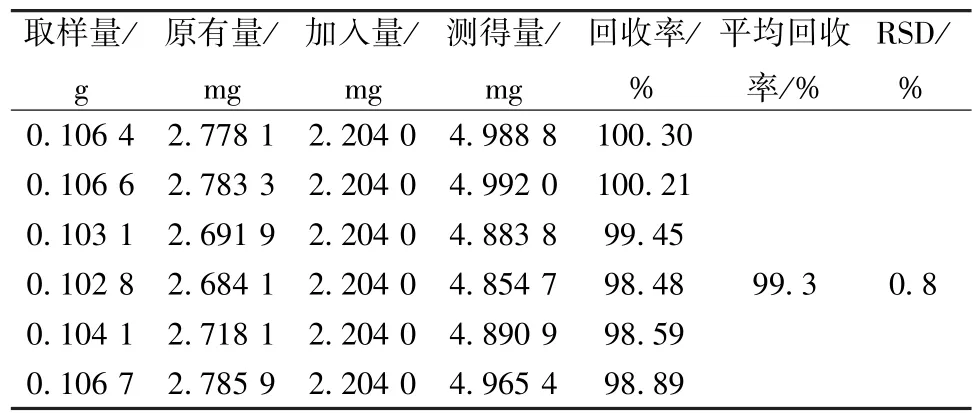
表6 朝藿定C 加样回收率试验结果Tab.6 Results of recovery tests for epimedin C
2.6.2.9 耐用性试验 取“2.6.2.7”项下供试品溶液、“2.6.2.2”项下对照品溶液,按“2.6.2.1”项下方法对不同色谱仪、色谱柱、检测波长、温度等条件进行考察,测得RSD 均小于2.4%,表明该方法耐用性良好。
2.6.2.10 样品含有量测定 取15 批样品,按“2.6.2.3”项下方法制备供试品溶液,取“2.6.2.2”项下对照品溶液,在“2.6.2.1”项色谱条件下测定,计算含有量(以干燥品计),结果见表5。8个品种15 批样品中朝藿定C 含有量为0.96%~5.95%,参照2015 年版《中国药典》 一部巫山淫羊藿含有量测定项下规定限度,建议将含有量限度暂定为按干燥品计算,含朝藿定C(C39H50O19)不得少于1.0%。
2.6.2.11 朝藿定C、淫羊藿苷含有量比较 不同品种淫羊藿根、叶中黄酮类成分含有量的差异很大;不同部位中淫羊藿苷含有量依次为叶高于根高于茎,但大部分品种根、叶中朝藿定C 含有量都较高[10-11]。取“2.6.2.3”项下供试品溶液,按2015 年版《中国药典》 一部淫羊藿项下方法测定淫羊藿根、朝藿定C 含有量,结果见表7,可知后者明显更高。
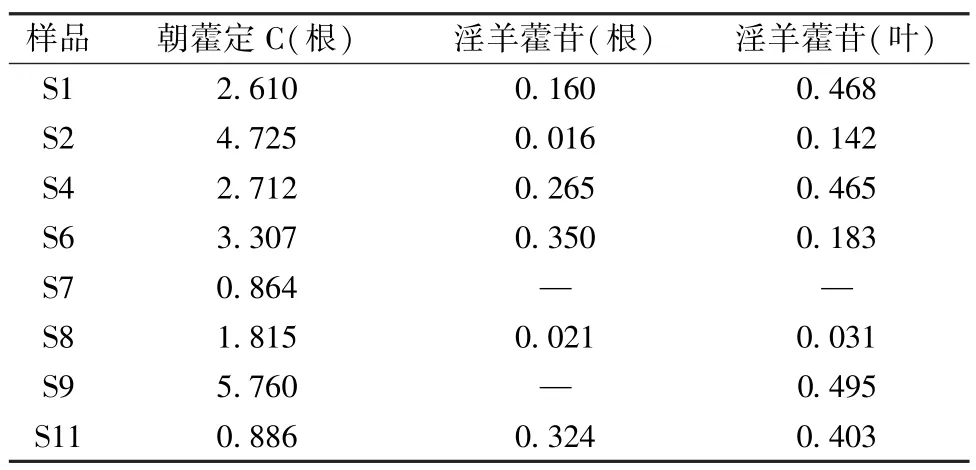
表7 各样品中朝藿定C、淫羊藿苷含有量比较(%)Tab.7 Comparison of contents of epmedin C and icariin in various samples(%)
3 讨论
黔岭淫羊藿、水城淫羊藿是贵州省独有品种,前者收载于2003 年版《贵州省中药材、民族药材质量标准》 黔淫羊藿项下(地上部分),是当地主流品种之一,蕴藏量较大[12-13]。因此,建议在淫羊藿根的来源中增加黔岭淫羊藿、水城淫羊藿根,以扩大贵州省该药材覆盖范围。
研究表明,淫羊藿主要活性成分为黄酮类,其中朝藿定A、朝藿定B、朝藿定C、淫羊藿苷、淫羊藿次苷含有量较高,5 种成分总量约占淫羊藿总黄酮的10%~20%[10]。2015 年版《中国药典》 一部淫羊藿(叶)以淫羊藿苷为指标成分(不得少于0.5%),巫山淫羊藿(叶)以朝霍定C 为指标成分测定含有量(不得少于1.0%)。本实验检测了8 批淫羊藿根中淫羊藿苷、朝霍定C 含有量,发现前者较低,均未达到《中国药典》 规定限度;后者较高,最低也有0.86%,与巫山淫羊藿含有量测定项下朝藿定C 规定限度相近,故以其为淫羊藿根的指标成分。
淫羊藿在加热炮制过程中,与朝藿定A、B、C、淫羊藿苷、宝藿苷Ⅰ母核相同的其他黄酮苷类成分可脱去糖基,转化为淫羊藿苷和宝藿苷Ⅰ[11],导致朝藿定C 含有量下降,淫羊藿苷含有量升高。因此,建议淫羊藿根药材洗净后晒干。
将淫羊藿苷作为淫羊藿的指标成分时,其含有量在某些品种中并不能达到《中国药典》 标准,易造成药材资源大量浪费[14-16]。因此,同时测定淫羊藿苷、朝藿定A、朝藿定B、朝藿定C 的总含有量作为淫羊藿质量控制标准较为合理,但其检验成本较高,有待进一步寻求更为理想的方法。
