取代基对2-(2-羟基苯基)苯并噻唑分子内氢键及质子转移的影响: 密度泛函理论研究
2020-04-28易平贵张志于陶洪文李洋洋彭文宇李玉茹
易平贵, 张志于, 陶洪文, 李洋洋, 李 庆, 彭文宇, 李玉茹
(湖南科技大学 化学化工学院 理论有机化学与功能分子教育部重点实验室 精细聚合物可控制制备及功能应用湖南省重点实验室 分子构效关系湖南省普通高校重点实验室, 湘潭411201)
1 引 言
质子转移反应普遍存在于化学与生物过程之中, 激发态质子转移(ESIPT)则是光物理和光化学过程中最基本的一种化学反应,近年来已受到了研究者的广泛关注[1~3]. 质子转移这种结构互变反应通常需借助其分子内氢键或分子间氢键传递质子或氢原子来实现,当在质子转移型分子的母体结构中引入不同取代基时,其作用会引起分子电子结构和电荷分布的变化,进而可改变氢键强度和质子转移等性质,因而基于质子转移的取代基效应是设计或改进质子转移型化合物功能的有效途径之一[4,5]. Palu-siak等[6]报道,当取代基在不同位置上时,异构体的能量会存在差异,质子转移过程所需跃过的能垒以及承受质子转移环上π电子的离域程度大不相同,其中最稳定的结构其芳香性最低,氢键强度最弱. Henary等[7]报道了溶剂化效应下不同取代基对激发态质子转移的影响,发现取代基效应和溶剂化效应等相互作用影响荧光性质.
2-(2-羟基苯基)苯并噻唑(HBT)分子结构简单且易于合成,是一种典型的具有激发态质子转移性质的有机分子,有关其ESIPT性质、机理及应用的实验与理论研究仍在持续报道[8~16]. 基于HBT基本结构及ESIPT性质的有机电至发光材料、光控分子开关及荧光分子探针等分子设计的研究也颇受关注. 此前,我们曾采用密度泛函理论(DFT)和含时密度泛函理论(TD DFT)并结合单重激发态相互作用法(CIS),通过对质子供体基团巯基的对位氢原子被取代后分子中苯环给出π电子能力变化的分析,探讨了气相中取代基电子效应对2-(2-巯苯基)苯并噁唑基态和激发态分子内质子转移的影响规律[17]. 在此基础上,本文采用DFT和TD DFT方法,选取典型的供电子基团羟基和典型的吸电子基团醛基,综合考察在二氯甲烷溶液中,取代基电子效应和取代位置对HBT质子转移赖以发生的分子内氢键、基态与激发态互变异构体相对稳定性、电子光谱以及质子转移能垒的影响规律. HBT及其衍生物的醇式(Enol)和酮式(Keto)两种互变异构体的分子结构如下图所示.

图1 HBT及其衍生物分子结构与质子转移Fig. 1 Molecular structures and proton transfers of HBT and its derivatives
2 计算方法
密度泛函方法已经成为研究分子体系电子结构、反应机理、有机化合物光谱性质的重要手段[18-20]. 经分析比较,发现含时密度泛函TD O3LYP[21]方法能很好地拟合HBT分子光谱的实验结果,本文分别采用密度泛函O3LYP和TD O3LYP结合6-311++G(d)基组及Tomasi[22, 23]等提出的可极化连续介质模型(Polarizable continuum model, PCM),对HBT及其衍生物分子在二氯甲烷(DCM)溶剂中的基态和激发态做几何结构优化和频率分析,以基态优化结构为基础用TD O3LYP/6-311++G(d)方法计算特征吸收光谱. 所有计算均在Gaussian09[24]软件包上完成.
3 结果与讨论
3.1 几何结构与氢键性质
以二氯甲烷为溶剂,对基态(S0)和激发态(S1)可能存在的结构进行全优化,结果表明HBT及其取代衍生物均存在稳定的醇式和酮式互变异构体, 它们的分子均为平面构型. 在S0态时,醇式比酮式构型稳定,而在S1态时,酮式则比醇式构型稳定. 氢键的结构与强度等性质会直接影响激发态质子转移反应,表1列出了分子发生质子转移的主要部位,即形成分子内氢键部分的键长、键角及两面角等结构参数,至于能量相对较高的反式醇式结构和反式酮式结构本文未予讨论. 可以看出,各分子的醇式结构从S0激发到S1态时,O5—H6键长增大,强度减弱,氢键N1…H6键长缩短,键角O5—H6…N1更接近180°,氢键强度增大;而酮式结构S0态的N1—H6键长则小于S1态相应的键长,键强度增大, 氢键O5…H6键长变长,键角O5…H6—N1进一步偏离180°,氢键强度减弱. 两种互变异构体氢键结构中键强度的变化均表明,HBT及其取代衍生物被光激发后,氢键性质的变化均促使激发态分子内质子转移的发生,即激发态比基态更有利于酮式结构的形成而发生质子转移. 从氢键的这种变化,还可看出取代基对HBT的ESIPT影响特点,相比于HBT,吸电子基团CHO的m-和p-位取代使S0态的O5—H6键长增大但S1态的O5—H6键长缩短, ESIPT的发生能力相对降低,而供电子基团OH的p-位取代有不利于ESIPT,m-位取代则有利于ESIPT. 可见,取代基效应对分子激发态质子转移的影响是通过其分子内氢键强度的变化而实现的[25],且这种影响是由取代基的电子效应和取代位置共同决定的.
表1 基态和激发态HBT及其衍生物优化结构的主要键长(10-10nm)、键角(°)和两面角(°)
Table 1 Theselected bond lengths (10-10nm), bond angles (°) and dihedral angles (°) of HBT andits derivatives in S0and S1states.
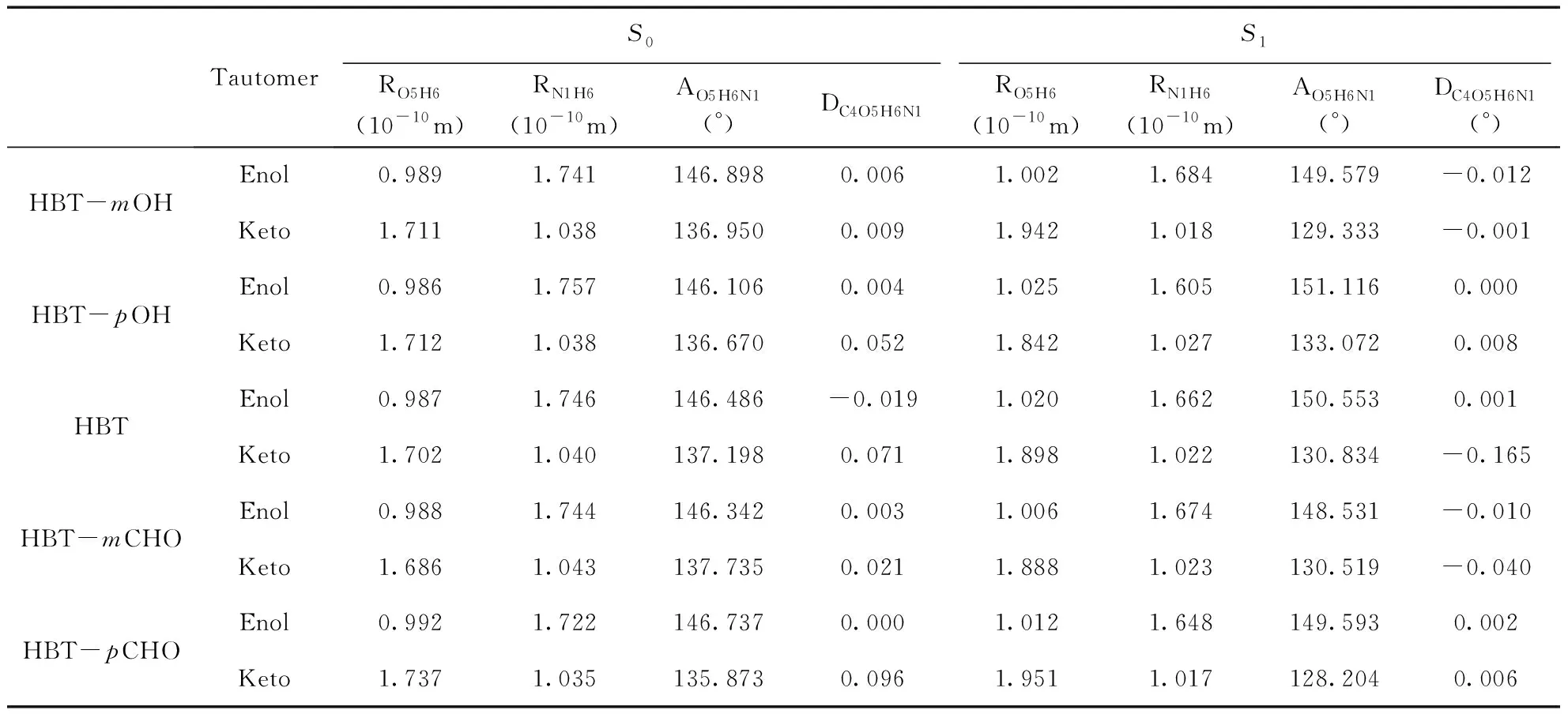
TautomerS0S1RO5H6(10-10m)RN1H6(10-10m)AO5H6N1(°)DC4O5H6N1RO5H6(10-10m)RN1H6(10-10m)AO5H6N1(°)DC4O5H6N1(°)HBT-mOHEnol0.9891.741146.8980.0061.0021.684149.579-0.012Keto1.7111.038136.9500.0091.9421.018129.333-0.001HBT-pOHEnol0.9861.757146.1060.0041.0251.605151.1160.000Keto1.7121.038136.6700.0521.8421.027133.0720.008HBTEnol0.9871.746146.486-0.0191.0201.662150.5530.001Keto1.7021.040137.1980.0711.8981.022130.834-0.165HBT-mCHOEnol0.9881.744146.3420.0031.0061.674148.531-0.010Keto1.6861.043137.7350.0211.8881.023130.519-0.040HBT-pCHOEnol0.9921.722146.7370.0001.0121.648149.5930.002Keto1.7371.035135.8730.0961.9511.017128.2040.006
另一方面,处于电子激发状态下的氢键动力学特性也可借助分子中特定化学键振动模式的光谱移动进行分析. 图2对比给出了HBT、HBT-OH及HBT-CHO基态和激发态Enol异构体分子内氢键中O1-H6键伸缩振动带的IR光谱图. 很明显,所考察的各体系从S0态到S1态,O5-H6键的伸缩振动峰均发生了较大幅度的红移,尤其是HBT-pOH的峰位红移最大(732 cm-1),从S0态的3281 cm-1到S1态的3549 cm-1,红移程度最小的则是HBT-mOH, 只有284 cm-1的红移. 此外,取代基对HBT这种O-H键伸缩振动的影响主要反映在S1态,对S0态不太明显. 按红移程度的相对大小,有利ESIPT发生的相对顺序为:HBT-pOH>HBT>HBT-pCHO>HBT-mCHO>HBT-mOH,显然与取代基的电子效应和取代位置有关,这与前面所讨论的氢键对HBT的ESIPT影响结论是一致的.
3.2 电子光谱与前线分子轨道
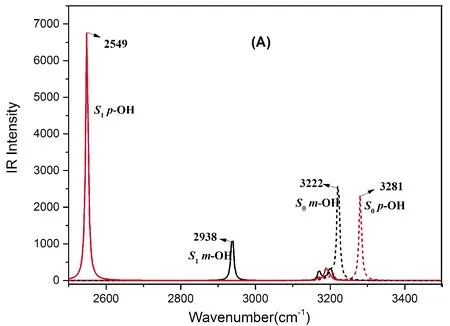

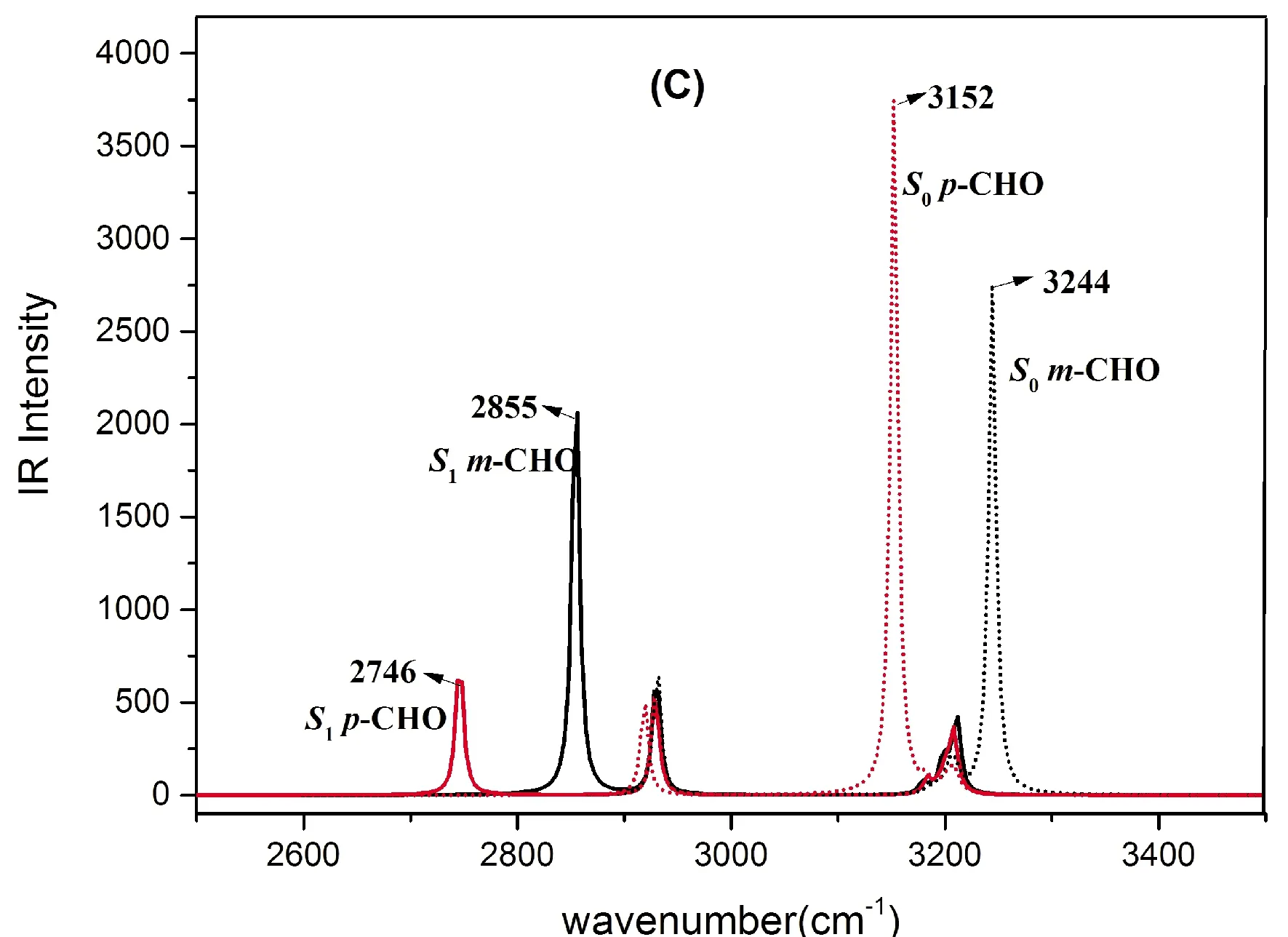
图2 基态和激发态HBT及其衍生物醇式构型O1-H6 伸缩振动计算IR光谱 (A) HBT-OH , (B) HBT, (C) HBT-CHOFig. 2 The calculated IR vibration spectra of enol form for HBT and its derivates in DCM solvent at the spectral region of O1-H6 stretching bands in S0 and S1 states. (A) HBT-OH , (B) HBT and (C) HBT-CHO.
化合物的光谱性质与分子的电子结构密切相关,为探讨目标化合物的电子跃迁机理,本文对HBT及其衍生物的基态和激发态的电子结构进行系统分析,应用TD O3LYP/6-311++G(d)/PCM理论方法模拟计算了它们在二氯甲烷溶液中的发射光谱和Enol构型的吸收光谱 (见图3),并在图4给出了基态Enol构型的前线分子轨道HOMO和LUMO示意图,表2列出了最大吸收波长λmax、发射波长λem、振子强度f及HOMO→LUMO跃迁对最大波长的贡献值CI. 对于HBT分子,醇式和酮式构型的最大荧光发射波长计算值分别为401和510 nm, 与实验值396和513 nm[15]非常接近. 可以发现,无论是最大吸收峰还是最大发射峰,都主要源于HOMO→LUMO的跃迁,除取代物HBT-pCHO吸收峰的CI值只有68.32%外,其它的贡献都大于95%. 各化合物的发射光谱相对于其吸收光谱发生了不同程度的红移,即Stokes位移,这是因为分子由基态向激发态跃迁时,分子的稳定构型发生了结构弛豫,分子在激发态下重新进行结构和电荷的布局,达到平衡后体系能量相对较低. 取代基的电子效应及取代位置对光谱波长及振子强度均有影响,相比于HBT,m-位的供电子基团羟基、p-位的吸电子基团醛基使各异构体的发射峰和吸收峰均产生了程度不大的蓝移,而p-位羟基、m-位醛基却使发射峰和吸收峰产生了明显的红移,两者发射峰的红移均超过了50 nm. 由此可知,取代基对质子转移型分子发光性质的影响,不只是由简单的取代基吸电子或供电子性质决定,与取代基所处位置也密切相关,有时甚至与取代基的立体相应也有很大关系,这些因素的综合影响有可能使分子在ESIPT过程中大幅度的结构扭曲而导致发光性质根本性的变化.


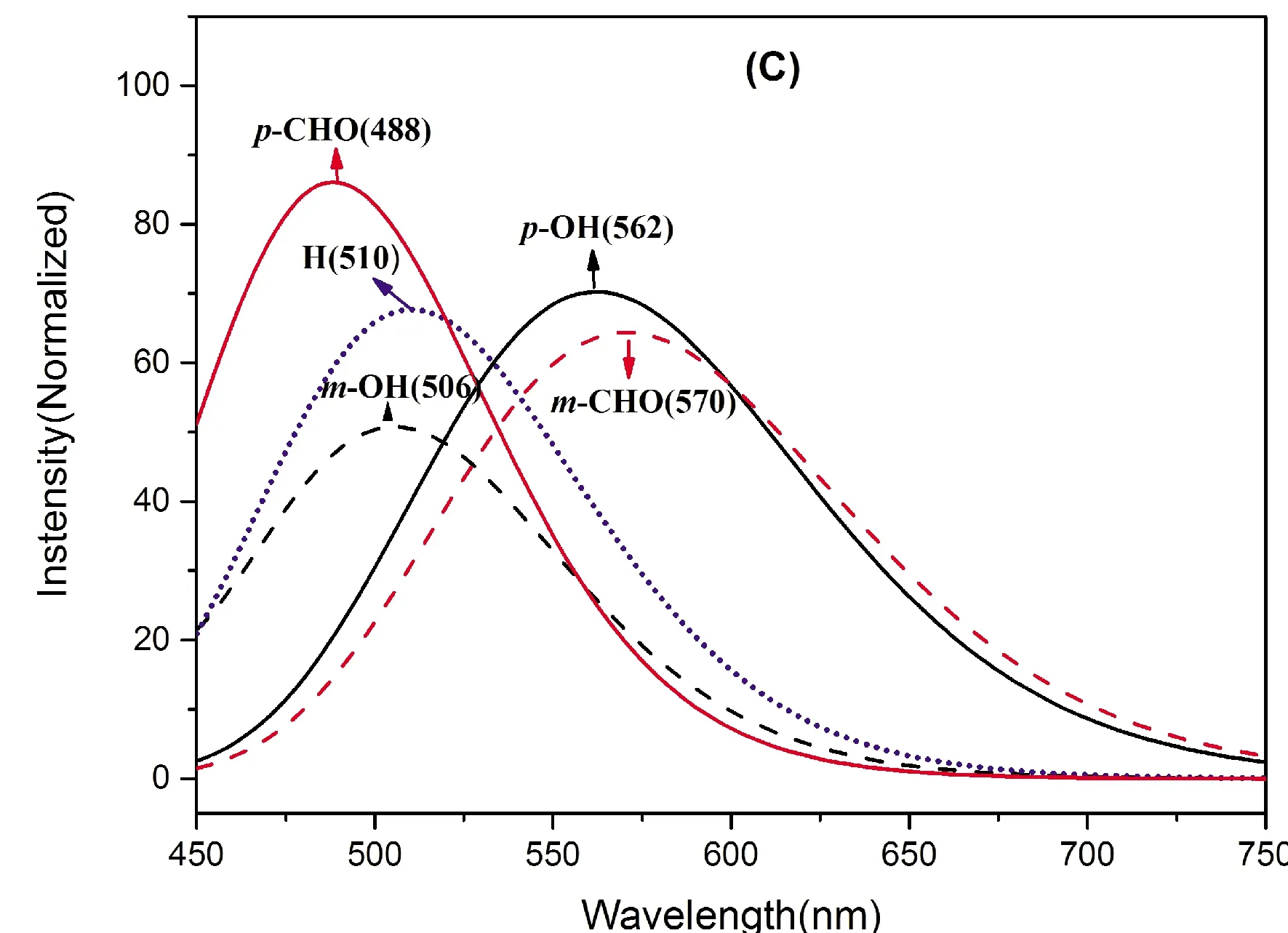
图3 DCM溶剂中HBT及其衍生物的计算光谱 (A) 醇式吸收光谱,(B) 醇式发射光谱,(C) 酮式发射光谱Fig. 3 The calculated absorption and emission spectra of HBT and its derivates in DCM solvent (A) enol: absorption , (B) enol: a emission and (C) keto: emission
由图4中容易看出,HBT及其衍生物的Enol构型的HOMO→LUMO跃迁即对应着π-π*跃迁,且LUMO轨道的羟基O5,即氢键的氢供体原子,电子云密度明显减少,但氢键的氢受体原子N1周围电子云密度著增加. 进一步从原子轨道对这两个前线轨道的组成贡献定量分析获知,对于HBT、HBT-mOH、HBT-pOH、HBT-mCHO及HBT-pCHO,O5原子对它们HOMO的贡献(%)依次为8.71、8.54、6.03、7.72和9.57,对LUMO为1.97、1.03、3.60、1.65和1.97;而原子N1贡献(%)则为2.91、2.51、2.89、3.98和1.98,以及10.17、6.61、8.85、9.70和10.04. 此外,轨道组成分析也可以得到取代基参与形成分子轨道的信息,取代基OH和CHO均参与了HBT分子前线分子轨道的共轭体系,如在HBT-mCHO中CHO对LUMO的轨道贡献为19.68%, HBT-pOH的HOMO中取代基OH贡献为8.27%. 这样,对于Enol构型,一旦发生HOMO → LUMO跃迁,激发态分子氢键中供氢原子O5去质子化能力增强,而受氢原子N1质子化能力增强. 对于激发态Keto,LUMO → HOMO的电子跃迁导致电子密度分布从含羟基的“酚环”向质子化杂环转移,从而增加了质子化杂环酸性与“酚环”的碱性,因此有利于其向基态衰变,进一步促进S0的Keto→Enol反向质子转移,最终完成ESIPT四级反应循环过程,即:Enol(S0)→Enol(S1)→Keto(S1)→Keto(S0)→Enol(S0).
表2 HBT及其衍生物的最大吸收和发射波长及相应振子强度(f)与HOMO至LUMO跃迁贡献率(CI)
Table 2 The maximum adsorption and emission wavelengths (nm), corresponding oscillator strengths(f) and transition compositions(CI) between HOMO and LUMO for HBT and its derivatives in DCM solvent

Adsorption(Enol)Emission(Enol)Emission (Keto)λmaxfCI(%)λemfCI λemfCI (%)HBT-mOH345.10.738195.12398.91.046998.59505.60.282399.23HBT-pOH391.70.297897.76462.20.374399.25562.20.3906100.00HBT348.80.468795.50401.40.725598.15510.30.3765.98.74HBT-mCHO396.50.544696.76458.40.846398.19570.60.3581100.00HBT-pCHO340.90.531368.32397.90.917095.80488.40.47879955
紫外吸收光谱的波长与S0时前线轨道的能量差有密切的联系,前线轨道能差越大表明吸收光谱的最大波长越短[26]. 图4中的HOMO与LUMO轨道能级差数据表明,取代基对HBT前线轨道能量差应综合其电子效应与取代位置来考虑,如吸电子基团CHO在HBT分子苯环羟基对位取代氢原子时,使HOMO与LUMO轨道能级差增大,吸收光谱较HBT为蓝移;而当其在间位取代氢原子时,HOMO与LUMO轨道能级差却减小,吸收光谱较HBT为红移.
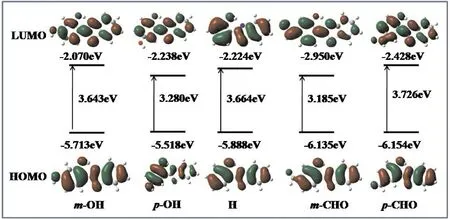
图4 DCM溶剂中HBT及其衍生物醇式构型前线分子轨道及能级Fig. 4 The frontier molecular orbitals and their energy levels of HBT and its derivatives in DCM solvent
3.3 质子转移机制与势能曲线
考察了两种取代基及其取代位置对HBT的分子质子转移过程能量的变化,按质子转移反应坐标方向逐步增大O—H键长做柔性扫描以获得它们的基态分子内质子转移(GSIPT)势能曲线和ESIPT势能曲线(图5). 可以看出,各分子基态的相对稳定构型为Enol,而激发态的稳定构型为Keto. 基态时, Enol构型的HBT-pOH能量比HBT-mOH高3.34 kcal/mol,Keto构型高4.48 kcal/mol;而在激发态时,则Enol构型的HBT-pOH能量比HBT-mOH高6.32 kcal/mol,Keto构型高3.12 kcal/mol. 吸电子基团CHO的取代衍生物相对稳定性的整体顺序正好与供电子基团的羟基取代物相反. 此外,随着O—H键长的逐渐增加,HBT及其衍生物在S0时能量呈现逐渐上升趋势,当O—H键长为0.1 nm左右时存在能量最小值,对应着Enol稳定构型. 随着质子转移的进行,各化合物由Enol向生成Keto的方向转变,由于芳香环遭到了破坏, Keto的能量明显升高,其结构性远小于Enol构型,故GSIPT是吸热过程,且质子转移的能垒很高. 由S1的势能曲线可以看出,各分子激发态Keto构型的能量明显低于Enol构型,ESIPT的发生后使体系更加稳定,由激发态的Enol生成Keto时只需跃过很小的能垒,ESIPT过程很容易进行. 综合S0的S1的势能曲线还可看出,基态的Keto转化为Enol构型,即基态的逆向质子转移,能垒非常小,有的几乎无需克服能垒;但在S1态时,逆向ESIPT过程,能垒却很高,难以发生. 因此, 按Enol(S0)→Enol(S1)→Keto(S1)→Keto(S0)→Enol(S0)方式进行的ESIPT反应,是热力学和动力学因素都为有利的自然过程. 取代基的电子效应与取代位置对GSIPT和ESIPT过程的能垒都影响不大,因此,从动力学角度看取代基效应对HBT质子转移影响不大.

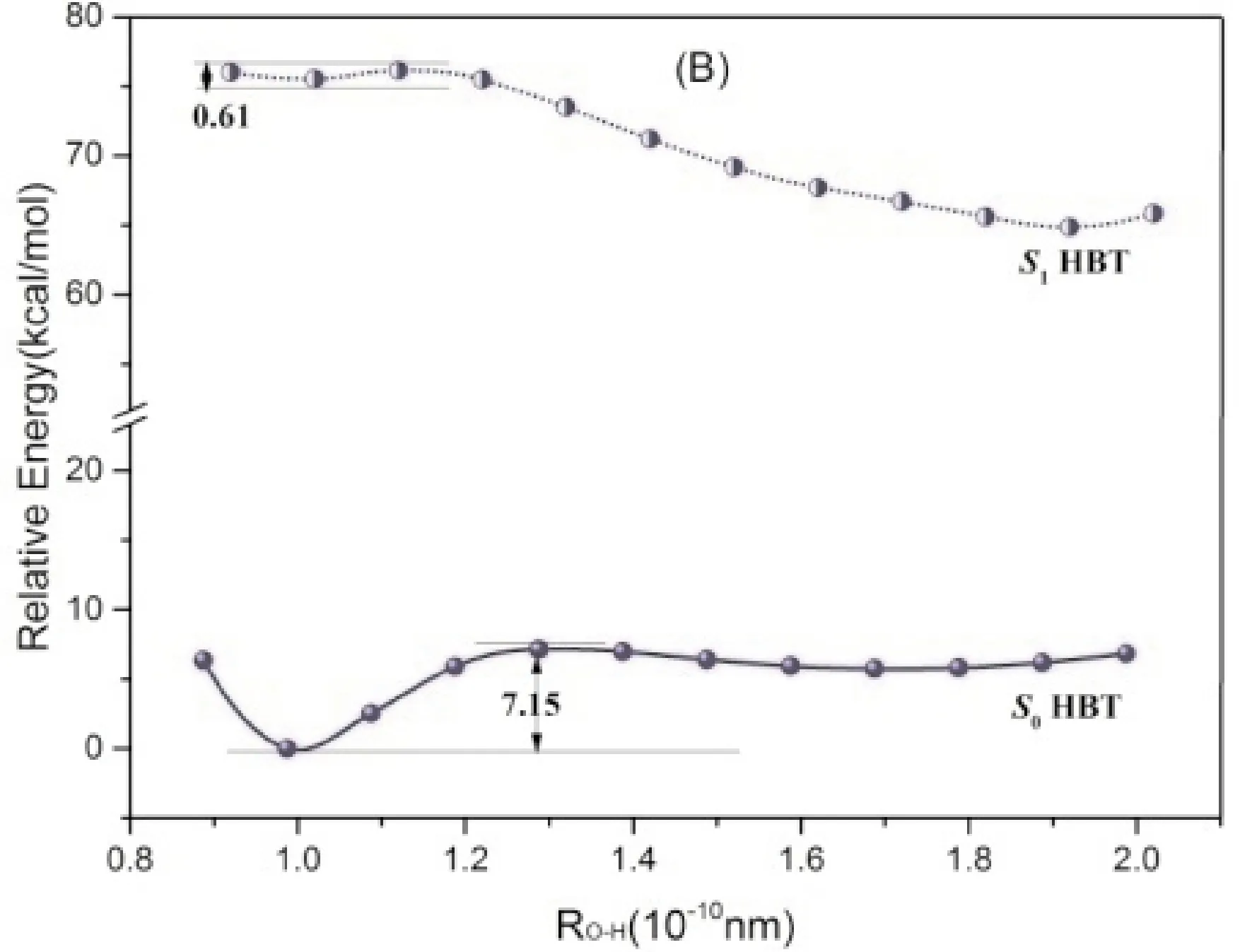
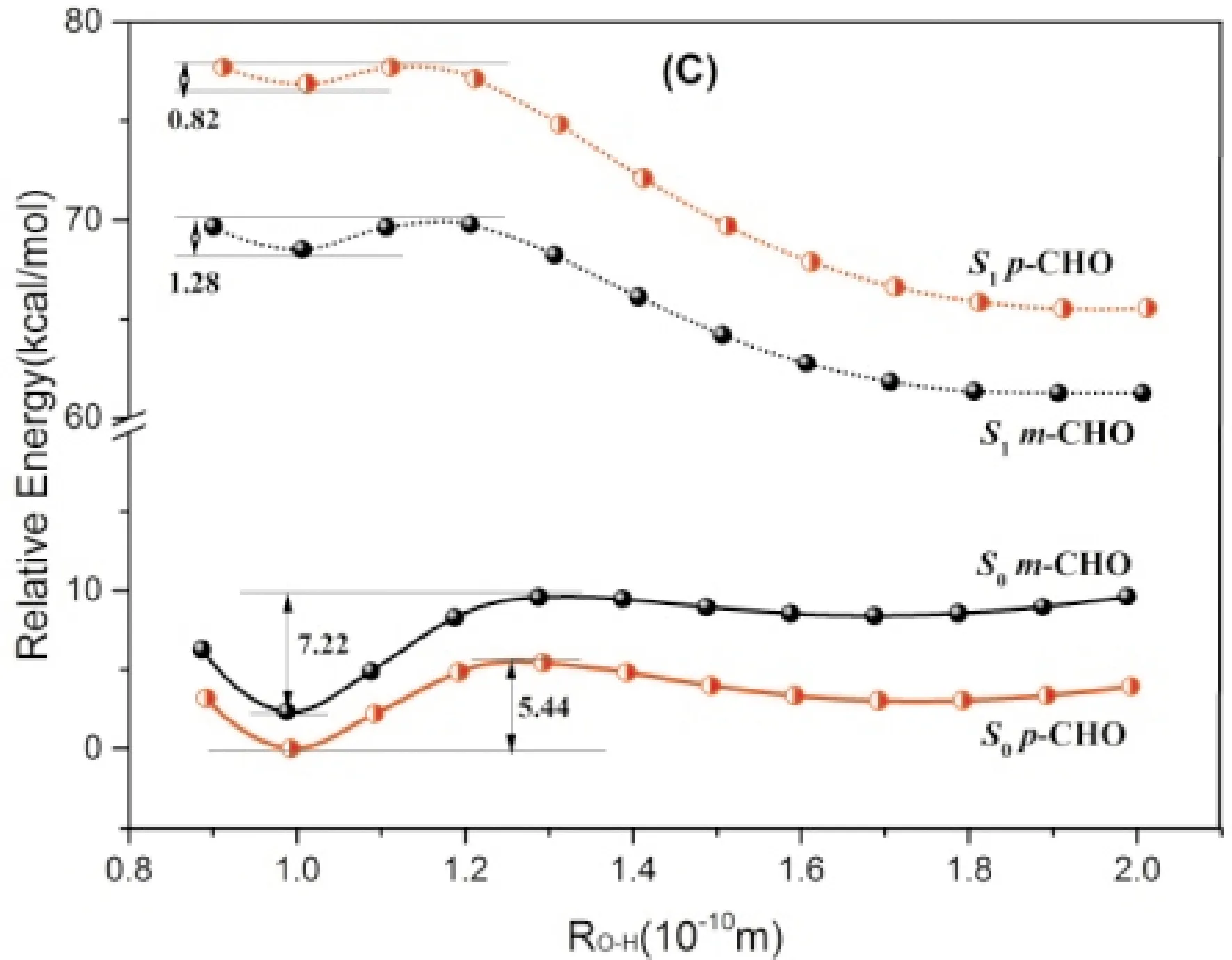
图5 DCM溶剂中HBT及其衍生物基态与激发态质子转移势能曲线Fig.5 Potential energy curves of proton transfer of S0 and S1 states for HBT and its derivatives in DCM solvent.
4 结 论
本文利用密度泛函O3LYP及含时密度泛函TD O3LYP考察了羟基和醛基对HBT的基态和激发态分子内质子转移的影响. 结果表明,HBT及其衍生物分子中酚环上的羟基氢可以和噻唑环上的氮形成分子内氢键,激发态时氢键强度增大. 基态时各分子以醇式构型稳定存在,而激发态的稳定结构为酮式, 取代基的电子效应和取代位置对基态和激发态异构体相对稳定性的影响是有差异的. HBT及其衍生物的最大吸收峰和发射峰主要源于前线分子轨道HOMO到LUMO之间的电子跃迁,光谱性质的变化既与取代基的吸或供电子性质有关,也与取代基所处位置有关. 基态分子内质子转移需要越过较高的能垒因而难以发生,而激发态时只需越过较低能垒就很容易发生激发态分子内质子转移.
