炎症小体在纤维化疾病中作用的研究进展
2020-04-26孙妩弋孙家昌杜佳佳李秀芹
李 南,孙妩弋,孙家昌,杜佳佳,李秀芹,魏 伟
(安徽医科大学临床药理研究所,抗炎免疫药物教育部重点实验室,抗炎免疫药物安徽省协同创新中心,安徽 合肥 230032)
炎症小体作为免疫系统的重要组成部分,通过直接识别病原体相关分子模式(pathogen-associated molecular patterns,PAMPs)和损伤相关分子模式(danger-associated molecular patterns,DAMPs),促进半胱天冬酶-1(caspase-1)和半胱天冬酶-11(caspase-11)分泌,是预防感染和组织损伤的第一道防线[1]。纤维化是由化学损伤、自身免疫反应、放射、过敏反应和持续感染等因素引起的慢性炎症反应的最后阶段,其特征在于细胞外基质(extracellular matrix,ECM)的过度沉积。纤维化最初是个可逆过程,然而,当纤维化程度加深时,可导致组织结构损伤和器官功能障碍,甚至引起癌症或死亡[2]。据统计,西方发达国家纤维化疾病造成的病死率高达总病死率的45%。目前,尚未有针对纤维化疾病治疗的有效方法,本文对炎症小体在纤维化疾病中的作用进行综述,为深入了解炎症小体在纤维化疾病中相关作用机制,以及寻找靶点促进药物的研究与开发提供参考。
1 炎症小体的结构
炎症小体通常是由1个模式识别受体(pattern recognition receptors,PRRs)作为传感器蛋白,通过凋亡斑点样蛋白(apoptosis-associated speck-like protein containing a CARD,ASC)与1个caspase组成的超分子组件[3](Fig 1)。其不仅在免疫细胞如单核巨噬细胞、T细胞中表达,在非免疫细胞如上皮细胞、肌成纤维细胞、角质形成细胞等同样有表达。目前研究较多的PRRs为核苷酸寡聚结构域样受体(nucleotide binding and oligomerization domain like receptors,NLRs)和黑色素瘤缺乏因子2样受体(absent in melanoma 2-like receptors,ALRs)[4]。NLR家族划分为5个亚型:含酸性反式激活结构域的NLRA、含杆状病毒抑制剂重复的NLRB、含半胱氨酸天冬氨酸蛋白酶募集结构域(caspase-activating and recruitment domain,CARD)的NLRC(NOD1、NOD2、NLRC3~5)、包含蛋白结构域的NLRP(NLRP1~14)、结构域未知的NLRX。ALR家族分为黑色素瘤缺乏因子2(absent in melanoma 2,AIM2)蛋白和γ干扰素诱导蛋白16(IFN-γ inducible protein 16,IFI16)蛋白,其特征结构域有HIN结构域和蛋白结构域(Pyrin domain,PYD),但AIM2蛋白中C端仅包含1个HIN结构域,而IFI-16蛋白C端包含2个HIN结构域。
ASC有两个结构域:CARD和PYD,这使它能够连接传感器蛋白与pro-caspase-1之间的相互作用[5]。Pro-caspase-1是半胱氨酸天冬氨酸蛋白酶家族的一员,当传感器蛋白和ASC之间的PYD相互连接,并且ASC与pro-caspase-1之间CARD相互结合时,两个相邻pro-caspase-1分子的紧密结合导致自我催化裂解产生P10和P20亚基,构成活化的caspase-1。caspase-1活化后,将白细胞介素1β前体(pro-interleukin-1β,pro-IL-1β)和白细胞介素18前体(pro-interleukin-18,pro-IL-18)切割为其活性形式,IL-1β和IL-18均为促炎细胞因子,对天然免疫系统和适应性免疫系统有广泛的影响。
2 炎症小体的调节
炎症反应是机体的一种保护性反应,但炎症小体活化引起的IL-1β和IL-18的成熟与释放,一旦失控会引发组织的过度损伤,引发疾病[6]。PAMPs(脂多糖、肽聚糖、腺病毒、细菌和酵母中的β-葡聚糖等)和DAMPs(三磷酸腺苷、胆固醇、脂肪酸、尿酸和溶酶体损伤等)均可识别PRRs,促进炎症小体的组装与激活。NLRP3炎症小体活化需要启动和激活两种信号:第一种信号为启动信号,启动过程中,细胞膜上的Toll样受体(Toll-like receptors,TLRs)识别PAMPs和DAMPs等刺激信号,通过下游NF-κB信号通路来诱导NLRP3、pro-IL-1β、pro-IL-18的转录和翻译;第二种信号为激活信号,NLRP3炎症小体的3个部分组装成稳定的复合体,分泌相应的促炎细胞因子引发炎症反应。目前研究发现,NLRP3炎症小体的激活主要通过钾离子外流、活性氧(reactive oxygen species,ROS)产生和溶酶体损伤释放组织蛋白酶B(Cathepsin B)3种模式[7]。AIM2蛋白可直接与病毒(如HBV)、细菌或宿主本身的双链DNA结合,促使AIM2炎症小体组装与激活[8](Fig 2)。
炎症小体也存在负性调节。炎症小体活化的同时伴随着自噬体的形成,通过募集自噬蛋白(p62蛋白、LC3B蛋白)降解炎症小体,起到直接抑制作用[9]。NLR家族可以识别细菌细胞壁的组分如肽聚糖,然后在细菌入侵部位引入自噬必需的适配蛋白Atg16L1,促进细菌进入自噬体,从而抑制炎症小体的激活和炎性因子的分泌。细胞因子或细胞与细胞之间的相互作用均可负向调控炎症小体活性,如CD4阳性效应细胞和记忆T细胞抑制NLRP1和NLRP3炎症小体介导的caspase-1激活和IL-1β分泌。只含有PYD或CARD结构域蛋白可以分别阻断ASC与传感器蛋白PYD端连接或阻断ASC与pro-caspase-1 CARD端的连接。
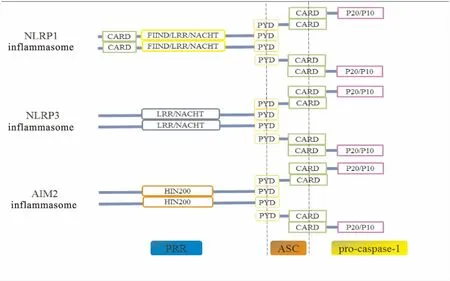
Fig 1 Domain structures of inflammasome proteins
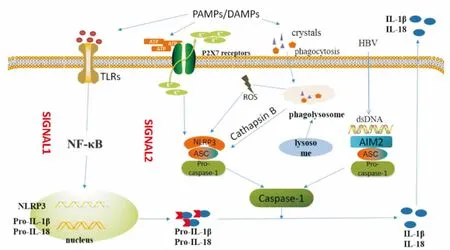
Fig 2 Mechanism of inflammasome activation
3 炎症小体与纤维化疾病
纤维化主要病理改变为器官内的纤维结缔组织增生和实质细胞减少。NLRP1、NLRP3、AIM2等炎症小体的激活在组织纤维化进程中起关键性作用,由炎症小体分泌的IL-1β、IL-18等促炎细胞因子可以加剧纤维化疾病的发展,最终导致器官结构破坏和功能减退。
3.1 肝纤维化肝星状细胞(hepatic stellate cell,HSC)活化后,转化为肌成纤维细胞导致ECM的沉积,形成肝纤维化。Lu等[10]用雄性BALB/c小鼠经腹部皮肤感染日本血吸虫,建立日本血吸虫肝纤维化模型,尾静脉注入腺相关病毒8构建NLRP3缺失小鼠模型,Western blot结果显示,与模型组相比,尾静脉注入腺相关病毒8组中胶原I和金属蛋白酶抑制剂-1蛋白的表达显著减少,表明肝纤维化与NLRP3炎症小体的形成与活化呈正相关。体外用日本血吸虫抗原(50 mg·L-1)处理HSC 24 h,然后用脾酪氨酸激酶(spleen tyrosine kinase,Syk)siRNA转染HSC。与对照组相比,血吸虫抗原刺激HSC引起Syk、p-Syk、caspase-1、ASC和NLRP3蛋白表达明显上升,转染Syk siRNA后明显抑制caspase-1、ASC和NLRP3的表达,结果表明体外血吸虫可能通过Syk介导的信号通路诱导NLRP3炎症小体的激活,促进Ⅰ型胶原和金属蛋白酶抑制剂-1蛋白表达,加剧纤维化发展。Inzaugarat等[11]选用雄性C57BL/6J野生型小鼠、NLRP3敲除鼠、NLRP3L351P/+Lrat Cre小鼠(NLRP3基因中第351位脯氨酸被亮氨酸替代后,获得NLRP3L351PneoR突变体小鼠,当NLRP3L351PneoR与Lrat Cre小鼠繁育后,仅限于HSCs表达NLRP3L351P的小鼠品系),腹腔注射脂多糖诱导NLRP3炎症小体激活。HE染色结果表明与模型组相比,NLRP3敲除鼠Ⅰ型胶原、α-SMA表达明显减少;NLRP3L351P/+Lrat Cre小鼠组出现窦周纤维化,巨噬细胞和中性粒细胞浸润无明显变化,免疫荧光结果显示分离的HSC中Ⅰ型胶原和α-SMA表达明显增强。外用LPS和ATP分别作为第一信号和第二信号,刺激野生型、NLRP3敲除型和NLRP3L351P/+Lrat Cre小鼠原代HSC。结果显示,NLRP3敲除组未表达IL-1β,而NLRP3L351P/+Lrat Cre小鼠α-SMA和转化生长因子β(transforming growth factor β,TGF-β)的mRNA水平相比野生型小鼠明显升高,推测NLRP3炎症小体在HSC的活化和肝纤维化发展过程中具有直接促进作用。
Zhang等[12]用BALB/c小鼠,经腹部皮肤感染15只血吸虫尾蚴建立肝纤维化模型,从感染当天(M0组)或感染后d 22(M4组)开始腹腔注射NLRP3选择性抑制剂MCC950(10 mg·kg-1),并维持注射到感染后d 56。Western blot结果显示,与对照组相比,模型组NF-κB p65蛋白明显升高;与模型组相比,M0组NLRP3蛋白和NF-κB p65蛋白表达明显降低,HE染色结果表明,肝纤维化程度有所改善;而M4组NLRP3蛋白和NF-κB p65蛋白表达与模型组相比无明显差别,肝纤维化程度均明显加重。体外选用人肝星状细胞系LX-2细胞,经日本血吸虫(10 mg·L-1)处理后,Western blot结果表明,与模型组相比,MCC950预处理4 h组NLRP3、α-SMA和胶原I的表达明显降低,说明日本血吸虫诱导LX-2细胞产生α-SMA和I型胶原依赖于NLRP3蛋白的表达。以上结果提示,NF-κB可能是NLRP3炎症小体的下游信号分子,促进肝纤维化发生与发展,而MCC950在肝纤维化初期具有预防作用,但在较晚时间点注射对于阻断炎症和抑制肝纤维化来说无明显作用。Mridha等[13]用雌性foz/foz和野生型C57BL/6J小鼠,喂养蛋氨酸/胆碱缺乏饲料建立非酒精性脂肪肝纤维化模型,并使用MCC950灌胃6周,结果显示与模型组相比,MCC950组中NLRP3、caspase-1、IL-1β和IL-6表达降低,纤维化程度减轻。体外使用胆固醇刺激枯否细胞和巨噬细胞,Western blot结果显示,与对照组相比,模型组释放IL-1β增加;与模型组相比,MCC950组抑制了IL-1β的释放,且减少了相关的中性粒细胞数。研究者认为MCC950可能通过巨噬细胞阻断的NLRP3炎症小体的激活,减轻非酒精性脂肪性肝炎造成的肝损伤。以上实验结果提示,无论是何种原因造成的肝纤维化模型,NLRP3炎症小体均促进肝纤维化发展,然而在肝脏不同细胞上的具体作用和表达差异,还需要进一步研究证明。
3.2 肾纤维化肾纤维化是各种慢性肾脏疾病发展的共同病变过程,主要病理改变为肾脏在受到内外多种致病因素的刺激后发生固有细胞损伤、大量胶原聚积和ECM的过度沉积,最终导致肾功能完全丧失。Gong等[14]用野生型C57BL/6J小鼠和NLRP3敲除小鼠,建立5/6肾切除术模型。与对照组相比,野生模型组小鼠肾脏ECM明显沉积,且胶原Ⅰ和胶原Ⅲ的表达与NLRP3蛋白表达呈正相关;与模型组相比,HE结果显示NLRP3敲除鼠肾小管间质纤维化显著减弱,ECM沉积明显减少。表明NLRP3对肾纤维化的形成起到促进作用。体外使用近端肾小管上皮细胞,转染NLRP3 siRNA,再用TGF-β1处理48 h,结果表明,沉默NLRP3可以抑制TGF-β1诱导的ROS水平的升高,降低线粒体膜电位水平。提示NLRP3炎症小体表达与线粒体功能和ROS水平相关。Sun等[15]用雄性C57BL/6J野生型和远端肾小管特异性ATG7基因敲除小鼠,于单侧输尿管结扎术d 7后,检测发现与对照组相比,远端小管特异性ATG7基因敲除组NF-κB、NLRP3、caspase-1和IL-1β含量明显升高,提示远端小管特异性ATG7基因与NLRP3炎症小体激活及其下游IL-1β的分泌有关,细胞自噬通过抑制NLRP3炎症小体激活从而减轻纤维化。Ling等[16]用雄性C57BL/6J小鼠,建立单侧输尿管结扎术诱导肾纤维化模型,肾脏 caspase-1、IL-1β和IL-18的含量异常增加,进一步证明在肾脏受损过程中NLRP3炎症小体的活化,同时引起内质网应激的CHOP蛋白表达上调。与模型组相比,皮下注射100 μg·kg-1胃饥饿素(肾保护作用)阻断内质网应激,NLRP3炎症小体各组分蛋白的表达降低,肾纤维化程度减轻。提示NLRP3炎症小体作为一个新的治疗肾纤维化靶点,通过抑制内质网应激降低NLRP3炎症小体的活化,从而改善肾纤维化的发展。
3.3 肺纤维化肺纤维化是由多种原因致肺组织损伤后的病理过程,早期表现为弥漫性肺炎,后期为成纤维细胞异常增生和ECM过度沉积。Sohn等[17]通过X-射线衍射仪照射雌性C57BL/6J小鼠左肺制备肺纤维化模型。Western blot结果显示,与对照组相比,模型组IL-1β、IL-4、IL-5、IL-6和IL-13促炎细胞因子水平与NLRP3炎症小体的表达呈正相关,免疫组化检测发现TGF-β1、IL-13和CD68(巨噬细胞标志蛋白)的水平特异性增加。推测辐射促进巨噬细胞中的NLRP3炎症小体激活,增加IL-1β释放,从而促进细胞因子(IL-4、IL-5、IL-6和IL-13)分泌,诱导肺炎和纤维化的发生。提示在肺组织中靶向抑制NLRP3炎症小体的活性,可以显著改善辐射诱导的纤维化,从而提高肺损伤治愈率。Sun等[18]研究了NADPH氧化酶对NLRP3炎症小体的影响,他们采用经口咽吸入多壁碳纳米管建立肺纤维化模型,结果显示NADPH氧化酶可以损伤溶酶体,组织蛋白酶B的释放增加,同时溶酶体损伤还会引起线粒体紊乱,包括线粒体外膜的通透性和ROS的生成,增加了NLRP3炎症小体生成。提示可以进行抗氧化治疗以减少NLRP3炎症小体激活,克服多壁碳纳米管引起的肺纤维化。
3.4 皮肤纤维化皮肤纤维化的特征是真皮层加厚以及一些附属物如毛囊、汗腺和皮肤血管的阻塞,普遍发生在系统性硬化症(systemic scleroderma,SSc)。SSc是一种病因不明的慢性特发性疾病,由活化的肌成纤维细胞介导的皮肤和内脏器官纤维化所致。Martinez-Godinez等[19]收集了21名局部性和弥漫性SSc患者皮肤样本,以及13名健康人皮肤样本。人内皮素-1已被证明参与皮肤纤维化的发展,研究者发现SSc患者皮肤中人内皮素-1的表达与NLRP3、IL-1β及IL-18表达呈正相关,提示SSc患者皮肤中的NLRP3炎症小体是纤维化和血管损伤的重要传感器,NLRP3炎症小体及相关的细胞因子激动剂或拮抗剂可作为限制皮肤纤维化的治疗靶点。Artlett等[20]体外用正常皮肤和SSc患者皮肤成纤维细胞株,扫描电镜结果表明,在使用caspase-1抑制剂后,细胞分泌胶原含量减少,qPCR结果显示NOD2、AIM2和NLRP3 mRNA表达水平均上升,但NLRP1 mRNA的表达并未增加,研究者认为这种差异可归因于NLRP1中基因位置,NLRP1可能存在功能的增强而不是其表达的增加。体内用野生型C57BL/6J、NLRP3基因敲除和ASC基因敲除小鼠,每日皮下注射100 μg博来霉素诱导皮肤纤维化,连续注射28 d。与模型组相比,NLRP3敲除组和ASC敲除组造模前后皮肤厚度无显著变化,提示NLRP3炎症小体及其下游信号分子可以作为治疗皮肤纤维化的有效靶点。
3.5 心肌纤维化心肌纤维化是由高血压等疾病导致的心肌成纤维细胞异常增殖,促使ECM过度沉积的病理过程。Wang等[21]用SD大鼠进行4周的高脂饮食诱导形成糖尿病模型,与对照组相比,糖尿病大鼠心肌ECM沉积增多;与模型组相比,AIM2基因敲除组ECM沉积较少,且胶原表达明显被抑制,证明抑制AIM2可以用于预防糖尿病所致的心肌纤维化。体外使用高糖处理H9c2心肌细胞6、12、24、48 h。与对照组相比,AIM2的表达与诱导时间的长短呈正相关;与模型组相比,抑制ROS可以降低AIM2的表达。提示高糖可以通过诱导AIM2炎症小体的产生,促使心肌纤维化发生,抑制ROS可以降低AIM2炎症小体活化,从而减缓心肌纤维化的发展。Pan等[22]用雄性C57BL/6J小鼠体内连续输注异丙肾上腺素(40 mg·kg-1)14 d建立心肌纤维化模型,与对照组相比,模型组小鼠体内NLRP3、TGF-β1、Smad表达增多,体外从C57BL/6J小鼠中分离原代心肌成纤维细胞,加入1 μmol·L-1血管紧张素II使其胶原表达异常增加,结果显示NLRP3、TGF-β1、Smad表达与刺激时间呈正相关。推测NLRP3-TGF-β1-Smad途径激活可导致心肌纤维化发生。Qiu等[23]采用SD大鼠结扎左冠状动脉前降支建立急性心肌梗死模型,造成心房间质纤维化。与对照组相比,模型组心房肌细胞横截面积比正常组明显增大,且上游调节因子硫氧还蛋白互作蛋白(thioredoxin interacting protein,TXNIP)与NLRP3、Pro-caspase-1、IL-1β、IL-18的表达呈正相关,推测TXNIP/NLRP3信号通路促进IL-1β和IL-18的成熟与分泌,并参与了心肌纤维化发生。以上结果提示,NLRP3炎症小体表达水平与心肌纤维化发生发展联系密切,且在不同信号通路中活性的调节还需要大量实验研究证明。
3.6 原发性骨髓纤维化原发性骨髓纤维化是一种原因不明的克隆性造血干细胞异常所致的慢性骨髓增生性疾病,又称原发性慢性骨髓纤维化。近年发现约50%慢性骨髓纤维化患者具有JAK2V617F突变,JAK2基因是一种胞内酪氨酸蛋白激酶,当第617密码子GTC第1位碱基由G变为T,导致其编码的缬氨酸变为苯丙氨酸。近年来有研究者[24]利用多能血液细胞系UT-7/GM建立了一种新的人血液细胞系D9,在加入四环素后表达JAK2V617F。与对照组相比,四环素刺激组AIM2蛋白表达与诱导时间呈正相关,推测AIM2是JAK2V617F在D9细胞中的下游信号分子,并通过AIM2炎症小体激活IL-1β的表达,从而造成骨髓纤维化。以上结果提示,AIM2炎症小体参与了携带JAK2V617F突变的患者骨髓纤维化发展。
4 展望
总之,自从Tschopp小组在2002年首次提出炎症小体这个术语,目前已经发现了二十多种炎症小体[25],近年来各炎症小体组分在纤维化疾病中表达异常增多,均说明炎症小体是各器官纤维化发生发展的重要环节。目前上市的抗纤维化药物极少,而且治疗效果不够理想,因此对于炎症小体相关信号通路的日益了解会为纤维化性疾病的治疗提供新的治疗思路与更直接有效的药物作用靶点。虽然很多学者利用NLRP3、AIM2等基因敲除鼠和基因沉默技术,可以更加有效地调控炎症小体的活性,但是仍面临许多问题亟待解决,如炎症小体与其他信号通路之间的相互关系,及炎症小体在不同疾病中具体激活途径等。此外,除本文提及的NLRP1、AIM2和NLRP3炎症小体外,其他炎症小体是否对纤维化疾病有影响,仍需进一步探究。
