非核糖体肽合成酶腺苷化结构域在E.coli中的表达及底物特异性分析
2020-04-23魏小雅于宏伟郭润芳
刘 欣,魏小雅,于宏伟,郭润芳
(河北农业大学 食品科技学院,河北 保定 071000)
微生物产生的一些次级代谢产物如杆菌肽、环孢菌素、博来霉素、细菌素和抗菌肽等,均有重要的药用价值和抑菌作用[1]。这些天然次级代谢物大多数属于非核糖体肽(Nonribosomal peptides,NRP),它们在细胞内的合成不同于常规蛋白质和多肽的合成,它们不需要核糖体作为翻译场所,也不需要信使mRNA作为模板,而且肽链中含有除20种天然氨基酸以外的稀有氨基酸[2],它们的合成由非核糖体肽合 成 酶(Nonribosomal peptide synthetase,NRPS)催化完成。NRPS是一个多酶复合物,大多数由3到15个模块构成[3],一个最小的NRPS模块有三个核心结构域,分别是氨基酸腺苷化结构域(A)、肽酰载体蛋白结构域(PCP)和缩合结构域(C)[4]。每个结构域执行单一化学反应以将单体单元结合或修饰成最终的天然产物。NRPS-A域在合成过程中起到正确的识别氨基酸,并且在消耗一个ATP后形成“氨酰-AMP”,同时释放一个焦磷酸,之后氨酰-AMP通过PCP结构域的作用形成“氨酰-S-PCP”结构的复合物,再由C结构域形成肽键,对肽链进行延伸[5],因此NRPS-A域决定了多肽中的氨基酸残基的上载。
抗菌肽(Brevibacillin,BBL)是侧孢短芽孢杆菌(Brevibacillus laterosporus)S62-9菌株产生的一种具广谱抑菌效果的小分子多肽,是由13个氨基酸残基组成的N端脂肪酸修饰的脂肽,其耐热性良好,121℃加热15 min依然具有良好的抑菌活性[6]。2016年张楠等[7-8]对BBL的遗传基因进行了定位,推测其遗传信息在质粒上,并推断出为非核糖体多肽。刘洋等[9]报道了其合成相关的基因簇,并确定了非核糖体肽合成酶的模块组成。BBL合成的第一步即为NRPS-A域特异性的去识别氨基酸并将其活化,因此了解NRPS-A域的功能研究对于阐明BBL的生物合成机制来说很重要,对于达到体外合成抗菌肽BBL是十分必要的。本研究将侧孢短芽孢杆菌(Brevibacillus laterosporus)S62-9菌株的质粒基因组中与BBL的相关的基因簇的NRPS-A域基因在大肠杆菌中异源表达,并获得了可溶的、有活性的NRPS-A域,以及检测了其特异性底物,进一步阐明了合成BBL的基因簇中的NRPS-A域在BBL合成过程中所起到的作用。
1 材料与方法
1.1 菌株和质粒载体
侧孢短芽孢杆菌S62-9菌株于本实验室保存、E.coliBL21购买于宝生物工程有限公司、E.coliRosetta(DE3) 和E.coliOrigami B(DE3) 购 买于北京博迈德基因技术有限公司、质粒pET-21b、pET-22b均为本实验室保存。
1.2 试剂
质粒小提取试剂盒购买于全式金生物技术有限公司,胶回收试剂盒购买于BIOMIGA(中国);Ni层析介质购买于北京博奥龙免疫技术有限公司;SDSPAGE试剂盒、Super DNA Marker、Es Taq MasterMix购买于北京康为世纪生物科技有限公司;NdeⅠ限制性内切酶、BamHⅠ限制性内切酶、NcoⅠ限制性内切酶、XhoⅠ限制性内切酶、T4 DNA Ligase、pMD18-T Vector购买于宝生物工程有限公司;焦磷酸检测试剂盒购买于Sigma生物试剂公司。
1.3 方法
1.3.1 质粒的提取 侧孢短芽孢杆菌S62-9质粒的提取方法参考文献[7]。
1.3.2 引物设计 根据NRPS-A基因序列和表达载体特征设计了4条引物(表1)。PS1、PS2、PS3均为上游引物,分别引入NdeⅠ、BamHⅠ和NcoⅠ限制性酶切位点;PA为下游引物,引入限制性酶切位点XhoⅠ。PS1/PA的扩增产物和pET-21b载体构建的重组载体中含His·tag基因,PS3/PA的扩增产物和pET-21b载体构建的重组载体有T7·ag基因,PS1/PA的扩增产物和pET-22b载体构建的重组载体中含有His·tag基因,PS3/PA的扩增产物与pET-22b载体构建的重组载体含有pelB leader基因。T7·tag可以增加融合蛋白的溶解性,His·tag用于融合蛋白的纯化;pelB leader可以帮助融合蛋白的分解到细胞外。

表1 PCR引物Table 1 PCR primers
1.3.3 NRPS-A结构域基因的PCR扩增 分别用3对引物扩增目的基因。扩增体系为:总体积25 μL、模板 1 μL、上下引物各 1 μL、2×EsTaqMasterMix 12.5 μL、双蒸水 9.5 μL。扩增条件为:变性 94 ℃1 min、退火61.5 ℃1 min、延伸72 ℃1.5 min。PCR扩增产物用1%的琼脂糖凝胶电泳分析,将目的条带按照胶回收试剂盒说明进行回收。
1.3.4 载体构建与序列分析 重组表达载体的构建方式见图1。将胶回收的PCR扩增产物分别与对应的线性化的表达载体连接,连接条件为16 ℃孵育12 h,具体添加量参见T4 DNA Ligase说明书。将重组克隆载体用42 ℃热激法转化至E.coliDH5α;挑取转化子进行菌液PCR验证,将验证正确的转化子提取质粒进行双酶切验证获得阳性转化子,具体步骤参照现代分子生物学试验指南[10]。将验证正确的阳性转化子送去测序公司测序,将测序正确的转化子保存测序结果进行生物信息学分析。

图1 重组载体构建示意图Fig.1 Schematic diagram of the recombinant vector
1.3.5 NRPS-A结构域基因的表达 将测序正确的重组表达载体pET21b-N1、pET21b-B2、pET22b-N2、pET22b-N3转 化 至E.coliBL21、E.coliRosetta(DE3)、E.coliOrigami B(DE3) 感受态细胞,筛选出12种阳性转化子。将阳性转化子接种于含有50 μg氨苄抗生素的LB培养基中37 ℃200 r/min摇床过夜培养,取过夜培养物按1%的接菌量转接到新的含有氨苄抗生的LB培养基中,37 ℃200 r/min摇床培养3~4 h,直至OD600为0.6~0.8时加入IPTG至终浓度为1 mmol/L,在20 ℃200 r/min诱导20 h。将诱导产物离心收集菌体,在冰浴条件下超声破(300 W,工作5 s,间歇10 s,共10 min)碎菌体,将破碎混合物12 000 r/min 4 ℃离心30 min,将沉淀和上清分离并用10%SDSPAGE进行检测,具体操作参考说明书。
1.3.6 可溶性融合蛋白的纯化 将诱导物离心收集菌体,将收集的菌体在冰浴条件下超声破碎(300 W,工作5 s,间歇10 s,共10 min)后离心收集上清,将上清液流经已经平衡好的Ni亲和层析柱,用10倍体积的咪唑浓度为0、20、40、80 mmol/L的洗脱液进行洗脱并收集洗脱液,将收集的洗脱液用10%SDS-PAGE分析。
1.3.7 氨基酸腺苷化活性测定 将纯化后的融合蛋白在ATP的推动下分别与20种氨基酸在37 ℃条件下作用15 min,反应结束后的20种反应物用焦磷酸检测试剂盒进行检测,具体添加量见说明书。此试剂盒标准检测限为PPi浓度在0.4 ~4 μmol/L,在此范围内焦磷酸浓度与荧光强度成正比,其线性方程为Y=11626X-1952。酶活(U)定义为每分钟产生1 μmol/L焦磷酸所用酶的量。
2 结果与分析
2.1 NRPS-A结构域基因的克隆
用PS1/PA、PS2/PA、PS3/PA作为引物,侧孢短芽孢杆菌S62-9的质粒DNA作为模板进行PCR扩增,结果如图2-1,泳道1、2、3分别为引物PS1/PA、PS2/PA和PS3/PA的扩增产物。PCR扩增后的到3条大小均为1.5 kb左右的条带,与预期大小一致,PCR产物依次命名为N1、B2、N3。4种重组表达载体的质粒双酶切验证结果如图2-2所示,泳道M为Super DNA Maker,泳道1~4分 别 是 pET21b-N1、pET21b-B2、pET22b-N1、pET22b-N3四种重组载体双酶切的电泳结果,每个泳道均检测到了2条条带,大小在1.5 kb的条带为NRPS-A基因5.5 kb左右的条带为线性化的表达载体此结果与预期结果一致。

图2 PCR扩增产物及质粒酶切结果Fig.2 PCR amplification product and plasmid digestion results
2.2 氨基酸序列分析
侧孢短芽孢杆菌S62-9的质粒基因组上非核糖体肽合成酶腺苷化结构域NRPS-A基因全长1.57 kb,共编码524个氨基酸,理论分子量为59.9 kDa,等电点pI为5.96。侧孢短芽孢杆菌S62-9的质粒基因组非核糖体肽合成酶腺苷化结构域NRPS-A与其他来源的NRPS-A同源性达54.2%(见图3),包含了典型的酰基活化酶(acyl-activating enzyme,AAE)的氨基酸序列TSGT(S)TG(图3下划线序列)以及6个AMP结合位点,分别为T169~S170、N309~ T314、I338、D405、R420和 K510。 另 外524个氨基酸中含有7个半胱氨酸,半胱氨酸易在蛋白折叠成高级结构时产生二硫键,这对于NRPS-A基因在E.coli中的可溶性表达带来一定的困难。该基因在Genbank中已注册,注册号为NO.MN218641。

图3 氨基酸的同源性比对结果Fig.3 Amino acid homology alignment
2.3 目的蛋白的诱导表达
2.3.1 不同表达受体的诱导表达 为了获得NRPS-A的可溶性表达,本试验尝试了构建不同表达 载 体(pET21b-N1、pET21b-B2、pET22b-N13和pET22b-N3)以及转化不同受体细胞(E.coliBL21、E.coliRosetta (DE3)、E.coliOrigami (DE3))进行表达分析,IPTG诱导表达后经SDS-PAGE电泳分析结果如图4。图4-1为4种表达载体在受体E.coliBL21中的表达情况,结果显示未出现与预期融合蛋白大小相近的条带,说明重组质粒在E.coliBL21中未表达;图4-2为4种表达载体在受体E.coliRosetta(DE3)中的表达情况,结果显示沉淀中出现与预期融合蛋白大小相近的条带,说明重组载体在E.coliRosetta(DE3)中出现了表达但是表达形式为包涵体;图4-3为4种表达载体在受体E.coliOrigami B(DE3)中的表达情况,结果均在沉淀中均出现了与融合蛋白大小相似的条带出现,而pET22b-N3在E.coliOrigami B(DE3)的诱导上清中出现了与融合蛋白大小相近的条带,其他3种在上清液中均未出现与融合蛋白大小相近的条带,说明只有pET22b-N3载体在E.coliOrigami B中出现了融合蛋白的可溶性表达。

图4 不同受体的诱导表达鉴定Fig.4 Identification of induced expression of different receptors
2.3.2 可溶性蛋白的纯化 将诱导的上清液用Ni亲和层析柱纯化,用咪唑浓度为0、20、40、80 mmol/L的洗脱液洗脱,将洗脱液用10%SDS-PAGE分析,结果显示含有40 mmol/L咪唑的洗脱液出现了大量的几乎且条带单一且大小和融合蛋白大小一样的蛋白,且活性检测程阳性,说明40 mmol/L咪唑的洗脱液洗脱下来的是目的融合蛋白。

图5 NRPS-A可溶性蛋白的纯化的SDS-PAGE分析Fig.5 SDS-PAGE analysis of purified NRPS-A soluble protein
2.4 特异性底物检测
收集有活性的洗脱液在有ATP推动的条件下在供试的20种氨基酸中筛选特异性的底物,结果发现融合蛋白对20种氨基酸有不同程度的腺苷化活性,对赖氨酸的腺苷化活性最大,并测定其酶活为4.66 U/mL,各氨基酸检测酶活如图6所示。
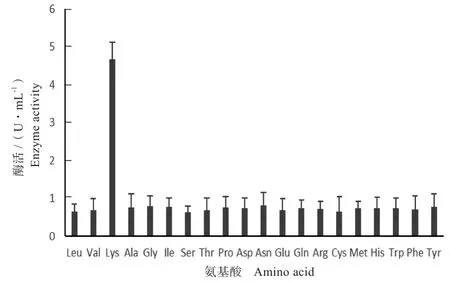
图6 特异性底物检测Fig.6 Specific substrate detection
3 结论与讨论
本试验构建的关于侧孢短芽孢杆菌S62-9的质粒中BBL合成基因簇中的NRPS-A基因的4个重组载体,在3个不同受体中的表达只有pET22b-N3这一重组表达载体在E.coliOrigami B中出现了可溶性表达,并且所表达的可溶性融合蛋白具有赖氨酸的腺苷化活性,这一研究结果为BBL的合成机制的研究提供了前提和保障。
NRPS-A域在NRP组装过程中起到决定性的因素,NRPS-A域底物特异性的研究是实现NRP体外生物合成的关键,长期以来一直是世界范围内学者普遍关注的问题,次生代谢产物大多数为非核糖体肽,就如本研究的侧孢短芽孢杆菌S62-9产生的抗菌肽BBL就是非核糖体肽,其中最核心的就是腺苷化结构域,BBL由5(Val、Leu、Ile、Lys、Tyr)种氨基酸构成的13肽,并且N端都是脂肪酸修饰链[9],值得一提的就是这五种氨基酸中就包括与本实验结果相对应的赖氨酸,NRPS-A域的赖氨酸特异性与BBL的氨基酸残基相对应,说明该结构域参与了赖氨酸的识别和上载,而其它氨基酸的识别可能由其它A域或其它机制完成。
研究NRPS-A对氨基酸的识别功能首先需要体外合成有腺苷化活性的酶蛋白,而在大肠杆菌中实现活性蛋白的异源表达有很多限制因素,其中表达载体的选择、表达载体构建、密码子偏好性以及受体的选择都是至关重要的[14]。根据这些限制因素我们做了多种尝试和改进,来自侧孢短芽孢杆菌S62-9的质粒关于合成BBL的基因簇中NRPS-A基因中存在多个E.coliBL21中不存在的稀有密码子,从而导致无法合成肽链[15],因此需要优化密码子或更换受体细胞才能解决这个问题,有研究表明大肠杆菌E.coliRosetta(DE3)菌株有稀有氨基酸所对应的tRNA,所以即使外源基因中含有稀有密码子也一样可以大量的表达[16]。继而选择E.coliRosetta(DE3)菌株作为受体进行表达。更换受体后,虽然4种表达载体都表达出了融合蛋白,但是融合蛋白都以包涵体形式存在,包涵体是一种不具有酶活性的形式,包涵体想要有活性,仍需要进一步的变性、复性等复杂步骤处理,导致了有活性的融合蛋白回收量很低[17]。通常认为包涵体的出现是由于目的蛋白表达速度快导致的,融合蛋白的折叠速度过快就会出现非正常折叠即包涵体形式,融合蛋白的高级结构正确与否是融合蛋白是否有活性的决定性因素之一[18]。因此有人采用降低培养温度和诱导剂浓度来降低基因的表达速率,而提高蛋白质的可溶性表达[19]。仔细分析目的基因NRPS-A发现,推导的氨基酸中有7个半胱氨酸残基,推测可能需要折叠形成二硫键才可能有活性,有研究发现E.coliOrigami B(DE3)菌株包含突变的硫氧蛋白还原酶和谷胱甘肽还原酶基因,可表达主要还原途径的两个关键酶,对含有二硫键的蛋白的表达有很大的帮助[20]。所以本研究更换了E.coliOrigami B(DE3)作为表达宿主,结果只有pET22b-N3这一重组表达载体表达出可溶的、有活性的蛋白,其他的融合蛋白均以包涵体形式存在,这一结果也说明受体中的一些还原酶对蛋白的活性表达来说也很重要,pET22b-N3重组载体中含有信号肽,信号肽的作用是可以更好的帮助融合蛋白分泌到细胞的周围空间或者胞外[21],本研究结果说明了适宜的载体和宿主有利于外源基因的可溶性表达。尽管有大量文献报道了外源基因在大肠杆菌受体中的表达研究,但具体到某个基因还需要具体分析。
