基于转录组测序对小麦抗叶锈性相关基因的筛选与分析
2020-04-23张艳俊杨毅清袁心悦王海燕刘大群
张艳俊,杨毅清,袁心悦,王海燕,刘大群
(1.河北农业大学 植物保护学院/河北省农作物病虫害生物防治工程技术研究中心/国家北方山区农业工程技术研究中心,河北 保定 071001;2.中国农业科学院 研究生院,北京 100081)
小麦叶锈病是由小麦叶锈菌(Puccinia tirticina)引起的真菌病害,是世界上分布范围最广的病害[1],其破坏性强,严重威胁到了小麦的生产安全。我国是小麦的生产、消费和贸易大国,其产量和消费量位居世界第一[2],同时也是世界上小麦叶锈病的流行区之一。2008-2009年,小麦叶锈病发生较为严重,2010年有所下降,2011年轻度发生,2012年河南、陕西、甘肃、四川、安徽等地小麦叶锈病大面积发生,2013年山东、河南和新疆局部地区发生严重,其他地区轻度发生[3-4]。2015和2016年在黄淮海地区爆发流行,且发病比往年约提前一个月[5-7]。目前,生产中主要依靠化学农药和抗病育种防治小麦叶锈病。化学防治虽然见效快,但是污染环境,不符合人、自然、社会和谐发展这一客观规律的要求,影响了生态文明建设,同时多次使用同种药物还会增加叶锈菌的抗药性。因此,种植抗病品种不但是防治小麦叶锈病最经济有效的措施,也是符合生态文明建设的要求,适合社会发展规律。
随着测序技术的发展,RNA测序(RNA-seq)通过提供基因表达的量化,为抗病候选基因的鉴定和分子标记开发提供了基础。RNA-seq是转录组学最强大的测序方法,它基于深度测序技术,可以比其他方法更精确地测量转录水平及其亚型[8],不需要基因组序列,即可分析寄主与病原物的转录本信息,从而使我们可以评估病原物是如何调节致病基因的表达以及如何影响寄主植物的防御反应[9]。Manickavelu等[10]通过对近等基因系小麦抗叶锈病与感叶锈病的cDNA文库测序,分析了Lr10相关的抗病基因表达情况。对抗病文库与感病文库中的ESTs进行对比,发现抗感反应间基因的表达存在差异。Kumar等[11]利用深度测序在小麦中鉴定了497个保守miRNAs和559个新的miRNAs,同时利用降解物组分析筛选到701个靶基因,并利用荧光定量PCR分析了miRNAs在与叶锈菌互作中的差异表达。张芳芳等[12]以RNA-seq测序数据为基础,筛选和鉴定了小麦LRRs类基因,又利用qRT-PCR技术对其进行了转录水平的表达分析,为深入研究小麦抗叶锈分子机制提供了新思路。
小麦抗叶锈病基因Lr19来源于长穗偃麦草,定位在7DL染色体上,为全生育期抗性基因,至今在我国很少发现对Lr19有毒性的菌株,具有很好的开发应用价值,是一个应用潜力很大的抗叶锈病基因。本实验室的张楠、王珅和高琳在小麦‘TcLr19’中克隆了TaPR1、TaPR2和TaTLP1等3个PR基因,并明确其表达受叶锈菌的诱导,利用基因枪法和VIGS技术对3个PR基因进行了功能验证[13-15]。本研究在对接菌后小麦材料TcLr19转录组测序的基础上,依据基因差异表达分析和基因功能注释结果筛选与抗叶锈菌相关的候选基因,采用qRT-PCR技术对RNA-seq数据进行了验证,为进一步鉴定和筛选抗病相关基因提供了可靠信息。
1 材料与方法
1.1 试验材料及处理
植物材料为小麦抗叶锈病近等基因系材料‘TcLr19’,其携带有小麦抗叶锈病基因Lr19,通过6代回交获得。
采用撒粉接种法接种叶锈菌07-10-421-3(FHJT)的夏孢子(孢子萌发率在80%以上)于一叶一心期的小麦材料‘TcLr19’上,分别剪取接菌0、1和6 d后的叶片,用液氮将其迅速冷冻,储藏于-80℃冰箱中,备用。
1.2 RNA-seq测序及原始数据处理、分析
将准备好的样品送上海派森诺生物科技股份有限公司进行转录组测序。基于Illumina NextSeq500测序平台,采用第2代测序技术(Next-Generation Sequencing,NGS)进行测序。用Sanger质量值Q来评估原始数据的测序质量,保证后续定量分析的准确性;利用Tophat2(http://tophat.cbcb.umd.edu/)将原始数据过滤后的Reads与已发表的中国春小麦基因组序列草图(the bread wheat cultivar Chinese Spring,IWGSC)[16]进行比对和定位分析,获得序列的来源基因以及表达产物的结构。
1.3 差异基因分析
利用HTSeq 0.6.1p2(http://www-uber.embl.de/users/anders/HTSeq)软件和RPKM(Reads Per Kilo bases per Million reads)方法,得出各样品中每一个基因的表达量,使用DESeq软件对基因表达进行差异分析。设置筛选两两比较组样本间显著差异表达基因条件为:|fold change| > 2,P-value < 0.05。将得到的差异基因进行GO功能富集分析和KEGG Pathway富集分析,以P-value≤0.05为阈值。
1.4 荧光定量PCR验证
选取差异表达基因,利用primer5.0软件设计引物(表1),以测序实验同批次采集样品的cDNA为模板,GAPDH(GenBank登录号:AF251217)为内参基因进行qRT-PCR。设3次生物学重复,应用LightCycler® 96荧光定量PCR仪进行扩增,采用2-△△Ct计算基因的相对表达量。

表1 用于qRT-PCR表达验证的基因及其引物Table 1 The qRT-PCR primers for the selected genes
2 结果与分析
2.1 转录组测序质量检测和RAW数据整理分析
通过对各样品的转录组测序数据的分析,分别获得Raw reads 48 258 292、47 346 190和56 341 268条,数据经过去除接头和质量过滤后,与原始数据相比,有效数据量均高于99%。各样品中GC含量均为58%以上,Q20值均高于98%(表2)。以上分析结果表明,各样品的RNA 样本测序所得原始数据的碱基组成基本平衡,且大部分reads 的碱基质量值大于20,符合数据质量错误率低于1%的要求,说明测序质量较好,数据具有可靠性。
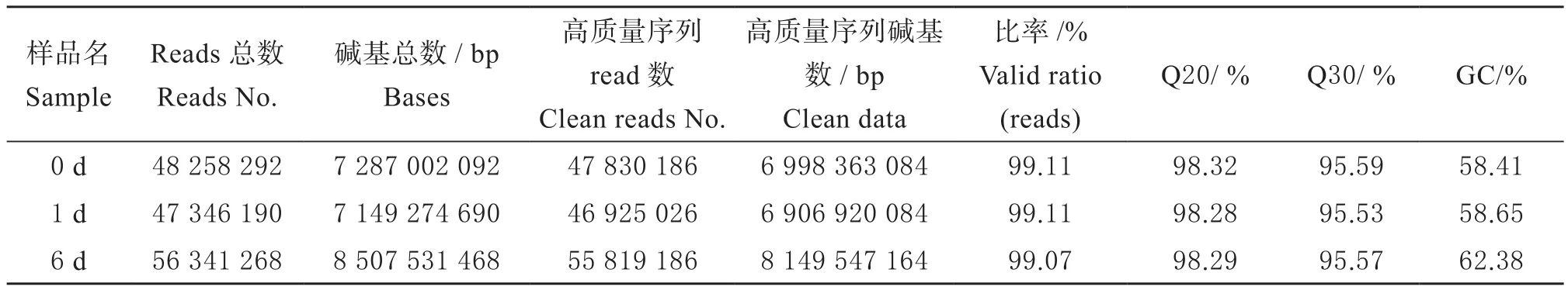
表2 数据统计Table 2 The statistics of data
2.2 基因比对分析
通过Bowtie2建立参考基因组索引,然后使用 Tophat2(http://tophat.cbcb.umd.edu/)将过滤后的Reads比对到参考基因组上。0、1和6 d样本比对上参考基因组的序列总数分别为34 275 708、33 853 145和37 746 006,比对上参考基因组的序列所占百分比分别为71.66%、72.14%和83.01%(表3)。以上分析结果表明,试验所产生的测序序列的Mapping比例都高于70%,Mapping成功,转录组测序参考基因组选择合适,且相关实验不存在污染。
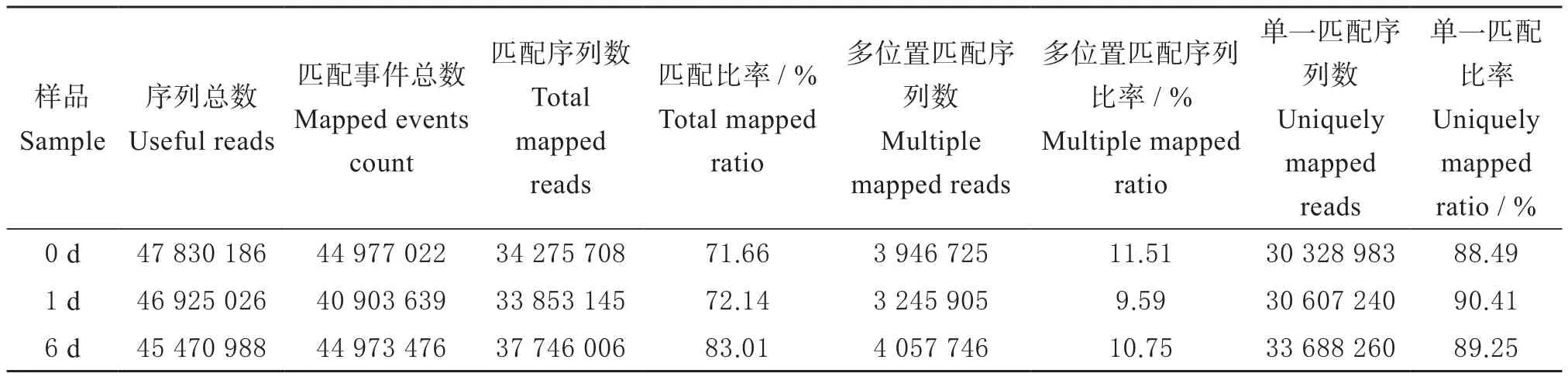
表3 RNA-seq Map统计Table 3 The statistics of RNA-seq Map
2.3 差异表达基因的筛选分析
利用DESeq软件,根据read count值分别筛选接种小麦叶锈菌后TcLr19在不同时间的差异表达基因。为了进一步获得与抗叶锈相关的抗病基因,对不同时间点数据的两两对比分析,在TcLr19中共筛选到差异表达基因8 970个,其中上调差异表达基因4 953个,下调差异表达基因4 017个。在1 d vs 0 d组合中差异基因最少,在6 d vs 1 d组合中差异基因最多(图1)。
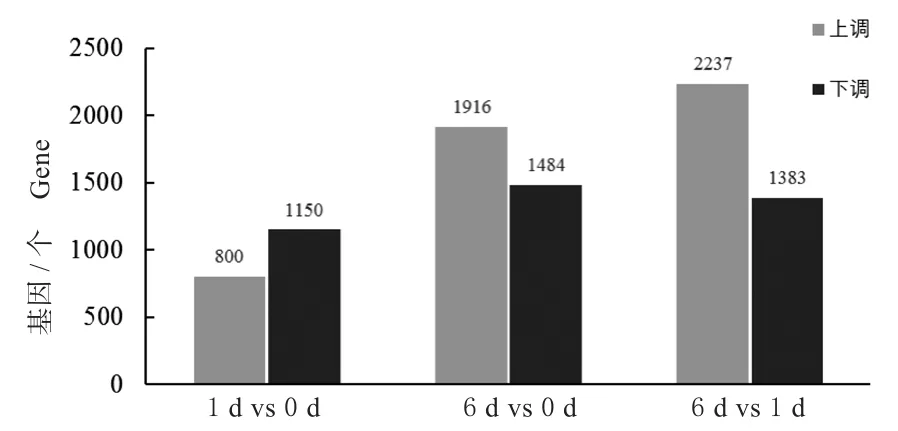
图1 不同侵染时间TcLr19的差异表达基因Fig.1 DEGs of TcLr19 with different infection time
2.4 差异表达基因的Gene Ontology富集分析
对差异基因进行GO富集分析,结果显示:差异基因被GO富集到分子功能、生物过程和细胞组分等3大类和1 790个小类中,其中分子功能部分包括489个、生物进程部分包括1 140个、细胞组分部分包括161个。选取GO富集基因中富集程度高和差异基因占比大的term作图(图2)。差异基因主要富集到催化活性和基础代谢term且富集程度高,6 d vs 1 d的富集term中差异基因数最多。
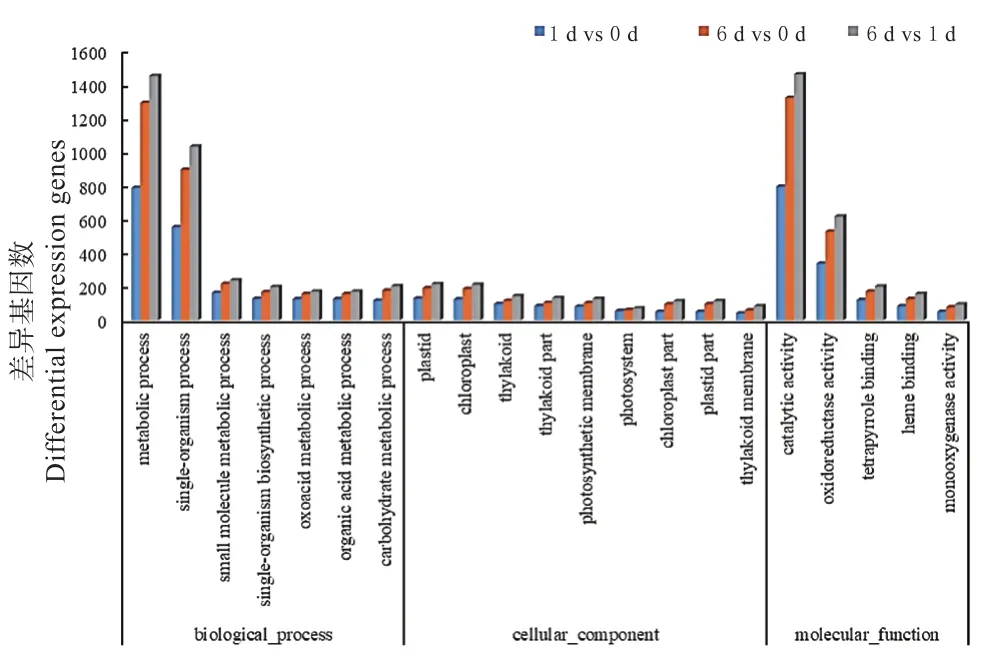
图2 差异表达基因的GO富集Fig.2 GO enrichment of DEGs
2.5 差异表达基因的KEGG富集分析
利用KEGG 数据库对上面已筛选出的差异基因进行了功能分类和Pathway 注释(表4),结果显示:3个时间对比组合中的差异基因定位到了84个代谢通路中,显著富集到了23个通路中,如氨基酸的生物合成(Biosynthesis of amino acids)、硫胺素代谢(Glycerolipid metabolism)、半胱氨酸蛋氨酸代谢(Cysteine and methionine metabolism)、甘氨酸、苏氨酸和丝氨酸代谢(Glycine, serine and threonine metabolism)等。

表4 差异基因KEGG富集分析Table 4 KEGG pathway enrichment of DEGs
2.6 差异基因Blast比对和在小麦与叶锈菌互作过程中的表达模式分析
通过以上差异基因的GO和KEGG分析,从转录组数据库中分别获得了Traes_5DL_D1D0CA79E、Traes_2AL_3D7807781、Traes_2BS_9931EE821和Traes_2BL_71ED6B5BD的序列。BLAST同源序列比对得知这些序列分别为NAC类转录因子、bZIP型转录因子、丝氨酸/苏氨酸激酶和酪氨酸蛋白激酶等与抗病相关的基因。又根据其序列设计qRTPCR引物,验证上面筛选出来的候选基因是否受叶锈菌的诱导及转录组分析结果的可靠性,结果显示:4个候选基因在0、1和6 d的表达量存在差异,且qRT-PCR与RNA-seq差异倍数分析结果虽在数值上不一致,但其差异基因的表达趋势与RNA-seq分析结果是一致的(图3),以上结果分析说明筛选出的差异基因受叶锈菌的诱导及利用RNA-seq技术分析基因表达的结果是可靠的。

图3 基因IDFig.3 Gene identify
3 讨论与结论
植物受到生物或者非生物的伤害时,植物体内的某些防御反应基因的表达就会发生变化,主要体现在时间、空间及产物含量的变化,从而起到抵抗外界物质的侵染及扩展的作用。本研究根据小麦近等基因系TcLr19与叶锈菌生理小种FHJT互作过程中可能会引起的变化,对接菌后0、1和6 d的样品,进行了 RNA-Seq测序。这与 Dobon等[17]采用RNA-Seq技术对条锈菌侵染小麦过程样品测序时间相似,能够彻底地记录这些防御调控基因在表达上的变化情况。Khalil-Ur-Rehman等[18]在RNA-seq分析的基础上,比较了夏芽(C1)、半休眠(C2)和内休眠(C3)时的赤霉素(GA)和脱落酸(ABA)相关转录本的水平,从而推测ABA和GA通路是调节葡萄芽休眠的调节开关。Yang等[19]对衰老后期的玉米叶片RNA-seq分析,发现了4 013个DEGs,且植物体内对氮运输和储藏起作用的天门冬酰胺合成酶(ZmAS3和ZmAS4)的2个基因也在干旱处理中被显著诱导,表明了在干旱诱导叶片衰老过程中,这些参与碳和氮代谢的基因表达谱发挥着调节作用。本研究通过对RNA-Seq数据的分析,获得差异基因8 970个,主要富集到了催化活性和代谢term中,参与到了硫胺素代谢、半胱氨酸蛋氨酸代谢和甘氨酸、苏氨酸和丝氨酸代谢等代谢通路,为抗叶锈基因的挖掘提供了大量的信息。
为了后续试验顺利进行下去,大部分研究利用qRT-PCR进一步确定RNA-Seq数据的可靠性。Liu等[20]在RNA-Seq分析基础上,利用qPCR验证套袋苹果梨4个成熟阶段转录组比较中的32个被鉴定为花青素生物合成途径基因的表达模式,结果与RNA-Seq结果高度吻合。其中,部分花青素生物合成通路基因MYB4-like1、MYB4-like2、MYB1R1、WDR在去袋9 d后表达上调,还发现了1 025个新基因,为揭示袋装处理苹果花青素生物合成机理提供了丰富的信息。Wang等[21]在全基因组高通量RNA测序的基础上,筛选出9个可能参与黄瓜性别分化调控的候选基因,有5个基因(Cs-MCM6、Cs-MCM2、Cs-CDC45、Cs-Dpri、Cs-CDC20) 参与细胞周期通路,推测细胞周期通路可能在黄瓜性别决定中发挥重要作用。Guan等[22]利用RNA-seq技术对冷处理前后的杂草稻(WR 03-35和WR 03-26)和栽培稻(Kongyu 131和9311)进行了测定,并对随机选取的12个DEGs进行半定量RT-PCR和qRT-PCR检测,结果发现这些基因半定量RT-PCR和qRT-PCR的表达模式与RNA-seq表达一致,进一步确定了RNA-seq数据的可靠性。本研究从RNA-seq数据库中筛选到了4个与抗病相关的候选基因(NAC类转录因子、bZIP型转录因子、丝氨酸/苏氨酸激酶和酪氨酸蛋白激酶),又利用qRTPCR验证了候选基因的表达模式,结果其受叶锈菌诱导且表达趋势与RNA-Seq结果一致,以上研究结果为获得小麦抗叶锈基因及其研究其抗病机制提供了丰富的信息。
通过RNA- seq技术对组织或胁迫诱导的差异表达基因分析,可以研究基因功能、代谢及调控途径等[23]。李芳等[24]在 RNA-seq基础上,通过BLAST同源序列比对和RACE技术克隆得到TaGSO1基因,并利用qPCR技术分析了TaGSO1基因在小麦与叶锈菌互作过程不同组合中的表达情况,推测TaGSO1基因参与了小麦抵抗叶锈菌侵染过程,为全面阐释小麦抗叶锈病机制奠定了基础。本研究下一步将结合RNA- seq数据的分析结果,筛选出与抗叶锈菌相关的候选基因,明确其与小麦叶锈病抗性的相关性,探索候选基因介导的小麦与叶锈菌互作的分子机制。
