噻唑与小分子间弱相互作用的理论研究
2020-04-22周芳芳王金树
周芳芳,王金树
(1.承德石油高等专科学校石油工程系,河北 承德 067000;2.吉林大学,吉林 长春 130012)
噻唑是一种含有硫、氮原子的五元芳香杂环化合物,分子上具有丰富的电子,因此能形成氢键、配位键及疏水效应等非共价相互作用,在生物制药、超分子识别过程及功能材料合成等众多领域具有广泛的潜在应用[1-3]。从理论上认识噻唑及其衍生物类分子与其他分子的相互作用的特征及本质特点,对噻唑及其衍生物的功能研究、超分子材料设计和合成具有重要的指导意义[4]。本文以噻唑与典型小分子形成的复合物体系为对象,采用高精度的量子化学手段,对其结构、能量、作用类型以及作用本质进行研究,从理论角度揭示含噻唑的超分子体系的结构特征和作用本质,进而为体系的设计和合成提供一定的指导。
1 计算方法
采用高精度量子化学方法,运用密度泛函理论(DFT),在B3LPY[5]/6-311+G**水平下,对NH3、H2O、HF 小分子与噻唑分子形成的复合物体系进行优化,并在相同水平下,通过频率验证,证明复合物结构为势能面上的稳定点。体系的相互作用能,定义为优化后的复合物体系的能量减去两单体的优化能量之和。采用“分子内原子”(AIM)理论,对弱相互作用的键关键点[即(3,-1)关键点]进行电荷密度拓扑分析,揭示弱相互作用的本质[6]。以上计算通过Gaussian09 程序包完成[7]。AIM 分析由Multiwfn 程序完成[8]。
2 结果与讨论
2.1 复合物结构与能量分析
为全面分析复合物结构,首先对噻唑分子进行表面静电势分析,结果如图1 所示。可以看出,噻唑分子平面内N 原子显示出非常强的负电性,H 原子则显示出明显的正电性,S 原子也显示出较弱的正电性。可以猜测,复合物结构中可能存在着两种氢键结构,一种为C-H∙∙∙N(O, F),另一种为N(O, F)-H∙∙∙N,因此选择了可能存在的稳定结构作为初始猜测结构进行优化,并得到势能面上的稳定结构,其优化结构如图2 所示。结构参数如列于表1 中。

图1 噻唑分子表面静电势/kcal·mol-1
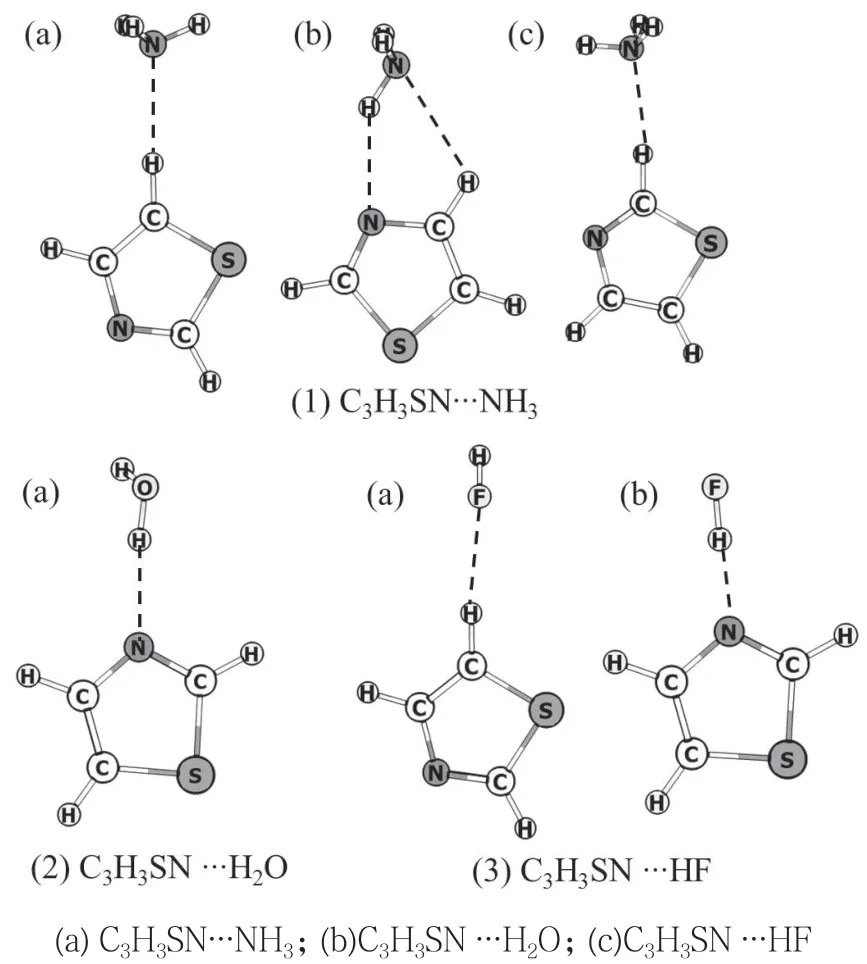
图2 噻唑与小分子形成复合物的稳定结构
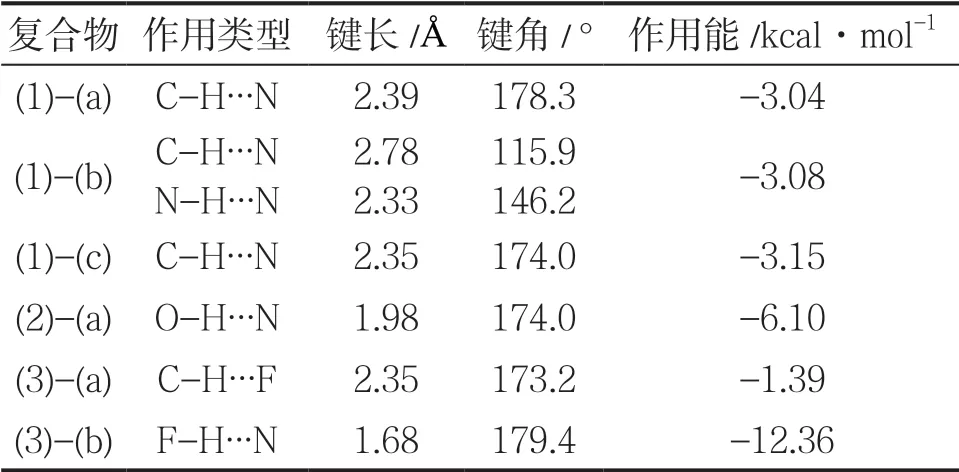
表1 噻唑与NH3、H2O 和HF 形成的复合物的结构参数
可以看出,噻唑分子与NH3可以形成3 种稳定的复合物。2 号的 C 原子和5 号的C 原子上所连接的H 原子正电性很强(图1),因此形成了2 个C-H∙∙∙N 氢键[(1)-(a)和(1)-(b)]。此外,N 原子的强负电性,使得噻唑分子与NH3形成了包含N-H ∙∙∙N和C-H ∙∙∙N 两种氢键的五元环状复合物结构。这3 种复合物的结构不同,但能量最大值[(1)-(c)]与最小值[(1)-(a)]仅相差0.11 kcal·mol-1。
噻唑分子与H2O 分子仅形成一种O-H∙∙∙N 氢键的稳定结构。与NH3分子相比,噻唑分子与H2O 分子形成的复合物的能量更低(-6.10 kcal·mol-1),这可能是H2O 分子中O 原子的强吸电子能力,使得与之连接的H 原子的电正性更高引起的。随着能量降低,分子间的作用距离更短。
噻唑分子与HF 分子可形成C-H∙∙∙F 和F-H∙∙∙N两种氢键的稳定结构。C-H∙∙∙F 氢键结构复合物的作用距离与C-H ∙∙∙N 氢键结构相近,但相互作用能仅为-1.39 kcal·mol-1,说明作为氢键接受体,F原子接受H 原子的能力比N 原子要弱得多。而在F-H∙∙∙N 氢键结构复合物中,相互作用能为-12.36 kcal·mol-1,比其他复合物的结构能量要低很多,且作用距离要短得多,表明F 原子的吸电子能力非常强,使得与F 相连的H 原子的电正性非常高,导致F-H∙∙∙N 氢键作用非常强。除五元环复合物结构外,所有结构中的氢键角度都接近于180°,表明氢键具有明显的方向性。
2.2 电荷密度拓扑分析
AIM 理论指出,利用复合物结构中氢键关键点[(3,-1)]处的电荷密度拓扑性质,可以对键的强度及键本质进行判断。可利用(3,-1)关键点处的电荷密度(ρ)的大小来判断作用的强弱,同时结合电荷密度的拉普拉斯量(∇2ρ)以及能量密度(H),来揭示弱相互作用的本质。各复合物结构中氢键关键点处的电荷密度性质如表2 所示。
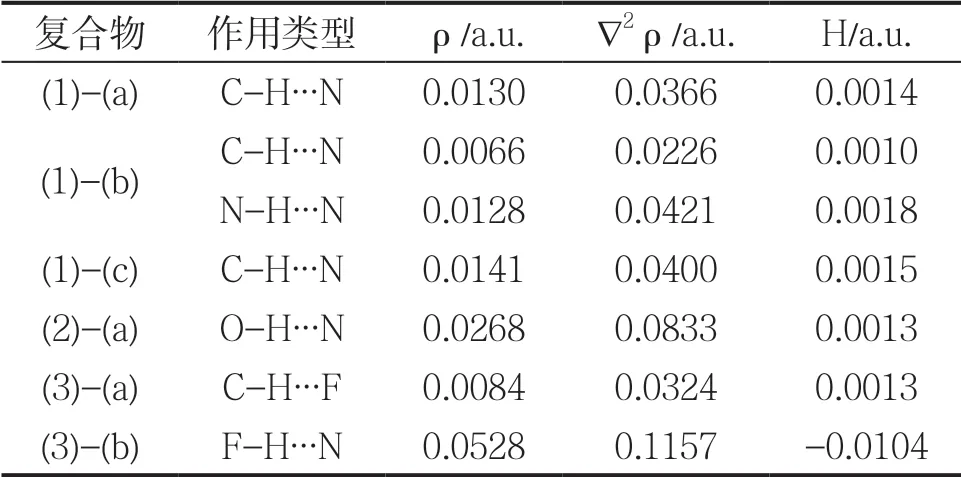
表2 复合物中氢键关键点处电荷密度性质
根据AIM 理论,氢键关键点处的电荷密度越大,键强越强。当电荷密度的拉普拉斯量和能量密度均大于0 时,此键属于纯闭壳层非共价相互作用;当拉普拉斯量大于0 而能量密度小于0 时,此键中含有一定量的共价成分。可以看出,复合物氢键关键点处的电荷密度变化与氢键强度一致;除F-H∙∙∙N氢键外,电荷密度值较小,说明氢键强度较弱,这与能量计算结果相一致。除F-H∙∙∙N 氢键外,所有复合物的氢键关键点处∇2ρ 和H 均大于0,表明这些氢键从本质上属于闭壳层非共价相互作用;而F-H∙∙∙N 氢键的∇2ρ 大于0,H 小于0,表明此氢键属于闭壳层作用,同时含有一定量的共价成分,这与非常强的分子间作用能力相一致。
3 结论
本文在B3LPY/6-311+G**水平上,计算了由噻唑分子与NH3、H2O 和FH 小分子形成的复合物的结构、能量及氢键关键点处的电荷密度拓扑性质。
从噻唑分子的分子表面静电势出发,对初始结构进行推测并进行优化和频率计算,得到势能面上的稳定点。计算结果表明,噻唑分子与小分子形成的均为氢键复合物,其中与NH3可以形成2 种C-H ∙∙∙N氢键复合物体系及C-H ∙∙∙N、N-H ∙∙∙N 氢键的五元环复合物体系;与H2O 仅可形成O-H ∙∙∙N 氢键体系。与HF 可形成2 种C-H ∙∙∙F 和F-H ∙∙∙N 氢键复合物体系。在能量上,从NH3、H2O 到FH,过程呈现逐渐增加趋势,除形成的五元环复合物外,其余氢键都具有明显的方向性。AIM 分析结果表明,除F-H∙∙∙N氢键外,氢键均为强度较弱的氢键,且本质是闭壳层非共价相互作用,F-H∙∙∙N 氢键在本质上含有一定量的共价成分。本研究可为噻唑类超分子功能材料体系的设计和合成提供一定的指导。
