基于高斯混合模型的肿瘤纯度估计
2020-04-21闫占正李玉双
闫占正,李玉双
(燕山大学理学院,河北秦皇岛066004)
高通量测序技术的快速发展为癌症基因组学研究带来了重大突破。在肿瘤与癌症研究中,在临床环境下获得的肿瘤固体组织中混合有正常细胞(即肿瘤不纯),使得筛选出来的癌症数据存在偏差,如果后续不经处理直接使用,往往会导致研究结果不理想甚至得到错误的结果[1]。 因此,进行肿瘤纯度的估计,即推测肿瘤组织中癌细胞的比例,已成为癌症研究中十分重要的课题[2-3],并已取得重要进展。如,ABSOLUTE 方法,利用单核苷酸多态性阵列数据(SNP array),借助最大似然估计计算肿瘤纯度[2];InfiniumPurify 模 型[4-6],利 用DNA 甲 基 化 芯 片 数据[7],将位点划分为超(次)甲基化位点,构建线性函数来估计肿瘤纯度;PurityEst,利用二代测序数据,通过对肿瘤样本中突变等位基因的比例建模来估计肿瘤纯度[8]。
与其他类型数据相比,DNA 甲基化数据能更稳定和灵活地显示肿瘤细胞和正常细胞之间的内在差异。观察正常样本,发现其甲基化水平分布与高斯分布相似,而有限高斯混合模型可以逼近任意概率分布密度函数[9]。因此,本文利用DNA 甲基化数据,借助高斯混合模型[10-11](Gmm, Gaussian mixture model)估计肿瘤纯度,得到的9 种类型的肿瘤纯度与ABSOLUTE 和InfiniumPurify 的结果高度一致。之所以与这2 种方法比较,是因为ABSOLUTE 是最早且最具影响力之一的纯度估计工具,常被用作评估的金标准,而InfiniumPurify 与GmmPurify 使用的数据相同,所求结果更具有可比性。
1 数据及其处理
癌症和肿瘤基因图谱TCGA 数据库(https://portal.gdc.cancer.gov/)中包含32 种肿瘤类型,每种类型都含有个数不同的肿瘤样本。包含正常样本的肿瘤类型有22 种,其中拥有2 个以上匹配的正常样本有21 种,多形性胶质母细胞瘤(GBM)只有1 个。由于多数肿瘤类型无(或只有很少)匹配的正常样本,本文选用文献[5]中构造的公共正常样本集进行操作,即在21 种肿瘤类型中每一个类型随机抽取2个正常样本,并与GBM 中的1 个正常样本一起组成包含43 个样本的公共正常样本集。每个肿瘤或正常样本均包含相同的485 577 个甲基化位点,其中大约9 万个位点的β 值全部缺失,有大约1 万个位点的β 值部分缺失(位点的β 值表示该位点的甲基化水平,0 ≤β ≤1,其中0 表示完全未甲基化,1 表示完全甲基化)。为避免缺失值带来的影响,将存在缺失值的位点全部舍弃。以包含466 个肿瘤样本和43 个公共正常样本的肺腺癌(lung adenocarcinoma,LUAD)为例,执行删除操作后剩余371 265 个位点。
2 GmmPurify 方法
2.1 高斯混合模型及应用
高斯混合模型[9]是指具有以下形式的概率分布模型:
这里μk是均值,σk是方差,αk是系数,φ(y|θk)是高斯分布密度,称其为第k 个高斯模型。每个高斯模型都是一个聚类。
当确定样本所属高斯模型时,可以利用最大似然概率算法来求解模型参数。设样本总数为N,样本集合为X={x1, x2, ..., xN}。设K 个高斯分布的样本数量分别为N1, N2, ..., NK, 第k (k∈{1,2,...,K})个高斯分布的样本集合为L(k)。则有

当无法确定样本所属模型时,可用期望最大化算法通过迭代进行求解,算法分2 步: E 步,根据参数θ(n)求 得 后 验 概 率P(xt|X;θ);M 步,更 新xt的 期 望E(xt),然 后 用E(xt)代 替xt求 出 新 的 模 型 参 数θ(n+1)。如此迭代直到θ 趋于稳定。
假设模型参数已知,随机初始化一组参数θ(0),可求得后验概率P(xt|X;θ)。求第k 个高斯分布,分别取x1, x2, ..., xN的概率,即求样本xt在第k 个高斯分布下的概率γ(t,k),有公式:

由式(4)~(7),可以推出:

随机初始化一组合适参数(α(0)k, μ(0)k, σ(0)k),k∈{1,2,...,K},将样本集合X 和初始化参数代入式(7)~(10),进行迭代直到参数趋于稳定,可求得样本集合X 的高斯混合模型参数(αk, μk, σk)。
将高斯混合模型应用于公共正常样本集,选取不同的K 值进行实验。实验需确保各类高斯模型存在的合理性以及各类模型之间明显的差异性。为此,制定以下实验条件:最小的分类概率不小于0.1,且各类之间的均值之差不小于0.1。研究发现,当K≤3 时,满足实验条件的甲基化位点占绝大多数。以LUAD 为例,其位点总数约37 万。当K=2 时,满足条件的位点约占29%;当K=3 时,满足条件的位点约占5%;当K=4 时,满足条件的位点约占0.4%。故设定K=3, N=43。以肺腺癌(LUAD)为例,依次处理371 265 个样本集合Xi={βi,1, βi,2, ...,βi,N}, i∈{1,2,...,371 265}, 其中βi,t表示第i 个位点在第t 个公共正常样本下的甲基化水平值。具体为:初始化参数αi,1=1/3,μi,1= min(Xi),σi,1=1,αi,2=1/3,μi,2= mean(Xi),σi,2=1,αi,3=1/3, μi,3= max(Xi),σi,3=1。将X1代入高斯混合模型,由式(7)~(10),迭代求得其解。迭代参数为:α1,1=0.450,μ1,1=0.033,σ1,1=0.006;α1,2=0.479,μ1,2=0.032,σ1,2=0.006;α1,3=0.071,μ1,3=0.303,σ1,3=0.149。依次计算,最终得到371 265 个位点在公共正常样本集中的高斯混合模型参数矩阵:

2.2 差异甲基化位点筛选
差异甲基化位点(DMPs)是指在肿瘤样本和正常样本中甲基化水平具有显著差异的位点,可提供肿瘤纯度估计的有效信息。非差异甲基化位点会成为噪音,从而削弱纯度信息[5]。 本文依据前面计算的公共正常样本集的高斯混合模型参数和位点在肿瘤样本中的甲基化水平值,通过定义重要的统计量来筛选差异甲基化位点。
定义1设某类型的肿瘤有m 个肿瘤样本,第i个位点在第j (j=1, 2, …, m)个肿瘤样本中的信息贡献率为

其中,
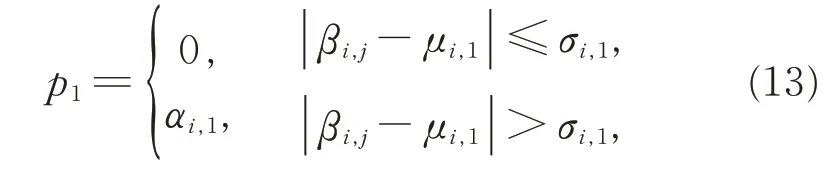
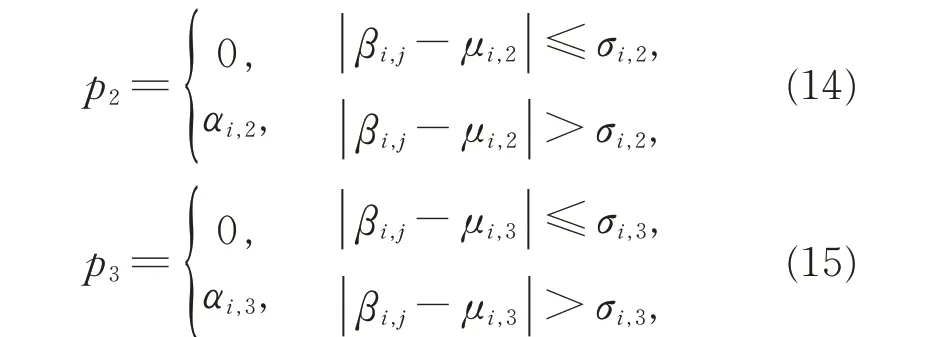
0≤Ri,j≤1,反映第i 个位点在第j 个肿瘤样本和公共正常样本中甲基化水平的差异程度。
定义2第i 个位点在肿瘤类型中的信息贡献值为

其中,0≤Si≤m,反映第i 个位点在肿瘤样本和公共正常样本中甲基化水平的差异程度,Si越大,说明差异越高。 将所有位点按Si非递增排序。设置阈值T,选取前T 个位点作为差异甲基化位点。
2.3 肿瘤纯度估计
如图1(a)所示,差异甲基化位点的β 值服从双峰分布[4],其峰值位置将用于肿瘤纯度估计。为提高差异甲基化位点的信噪比,采用文献[4]中的方法,先将双峰分布变为单峰分布:将差异甲基化位点划分为超甲基化位点和次甲基化位点(若位点在肿瘤样本中的甲基化水平均值高于正常样本中的甲基化水平均值,则称该位点是超甲基化位点;反之,称为次甲基化位点),依据合理性假设——超甲基化位点与次甲基化位点的β 值之和等于1,将次甲基化位点的β 变为1-β, 超甲基化位点的β 保持不变。对所有变换后的β 值进行核密度估计,其峰值对应位置的β 值即为肿瘤纯度。
3 结果及分析
由于GmmPurify 与ABSOLUTE 采用的数据类型不同,为方便比较,选取来自9 种不同肿瘤类型的3 759 个肿瘤样本,它们同时具有SNP array 和DNA 甲基化2 种数据,并对不同的肿瘤类型和比较对象(ABSOLUTE 和InfiniumPurify)设置了不同的阈值T。结果表明,GmmPurify 计算得到的纯度估值与 2 种方法的结果具有强相关性,和InfiniumPurify 的结果高度一致(见表1)。图1(b)展示了GmmPurify 和ABSOLUTE 对196 个肺腺癌肿瘤样本的纯度估值的强相关性(皮尔逊相关系数为0.834);图1(c)展示了GmmPurify 和InfiniumPurify对466 个肺腺癌肿瘤样本的纯度估值的强相关性(皮尔逊相关系数为0.929)。
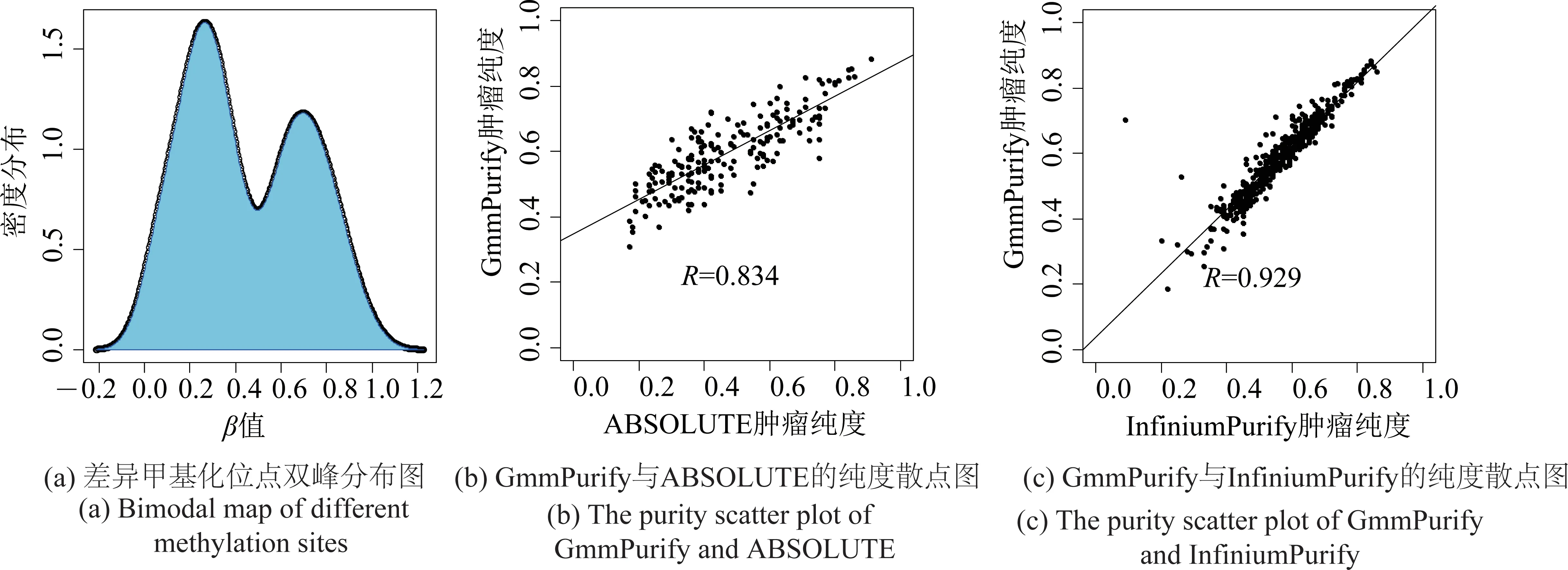
图1 肺腺癌(LUAD)选择T=200 时的结果展示Fig.1 The results of lung adenocarcinoma(LUAD)with T=200
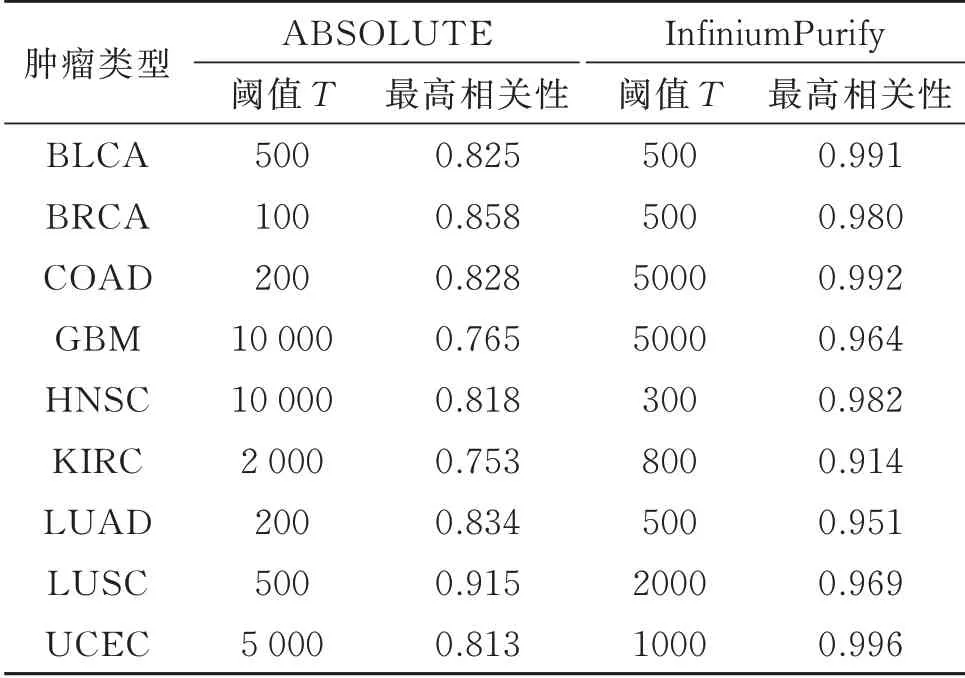
表1 最高皮尔逊相关系数表Table 1 Table of the highest Pearson correlations
此外,本文还测试了阈值T 对GmmPurify 纯度估计的影响。如图2 所示,T 过大或过小都会影响纯度估计的准确性。 如当T=50 000 时,GmmPurify 的 纯 度 估 值 与 ABSOLUTE 和InfiniumPurify 的相关性大大降低。可见,每种肿瘤类型都有各自的最佳阈值区间,在此区间内得到的纯度估值相对最优且比较稳定。如果要求所有的肿瘤类型都选择相同的阈值,建议T=1 000,此时能够得到令人满意的纯度估计结果(见表2)。
以上分析说明,GmmPurify 可以作为肿瘤纯度估计的可选择工具。
4 结 论
肿瘤纯度反映了癌症类型的内在特性以及临床收集样品的精确度,对后续的癌症数据分析有重要影响。本文提出了一种计算简单、可适用于多种肿瘤类型、具有较强生物学意义的肿瘤纯度估算方法GmmPurify。估算结果与经典的ABSOLUTE 和InfiniumPurify 高度一致,验证了方法的可行性。此外,所提出的统计量“信息贡献值”可作为后续研究DNA 差异甲基化分析的重要工具。
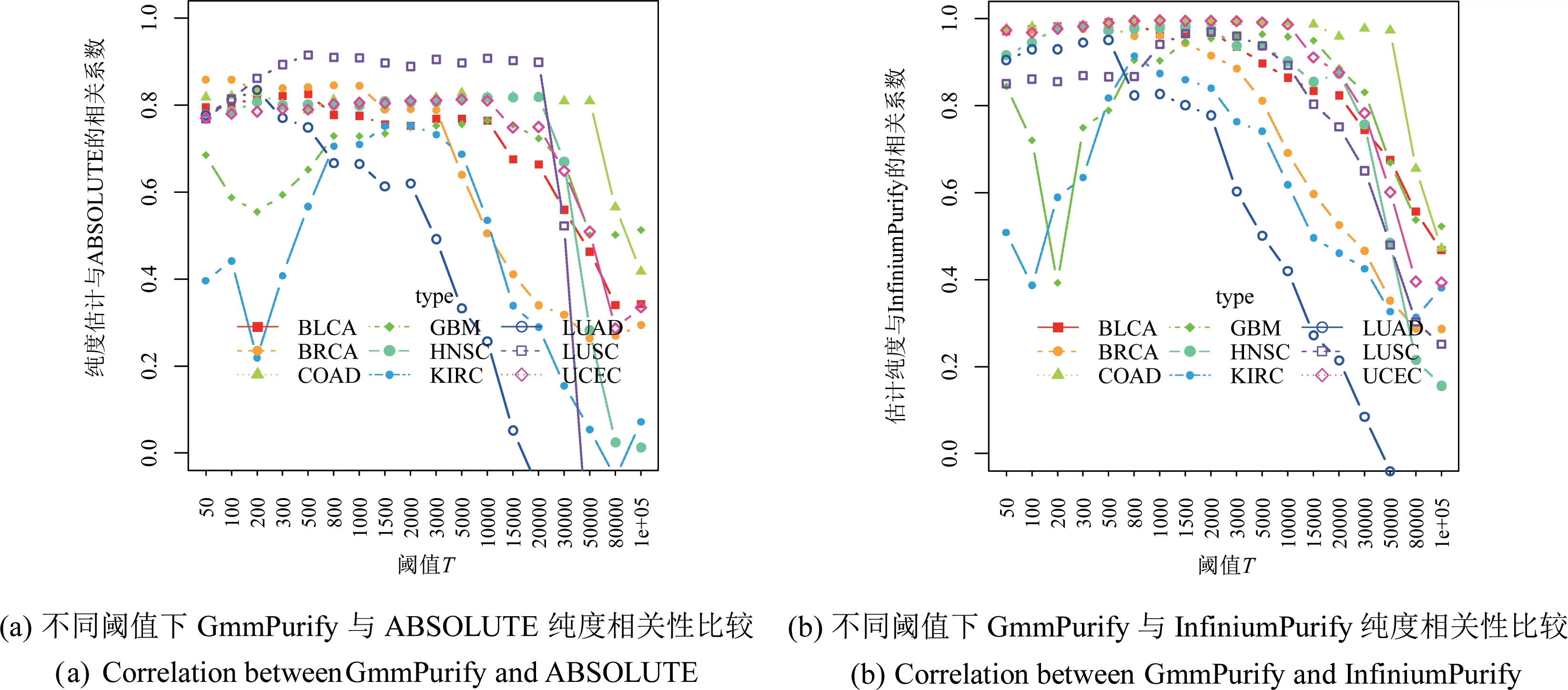
图2 9 种肿瘤类型在不同阈值下的纯度估值分析Fig.2 The analysis of purity estimations for 9 tumors with different thresholds
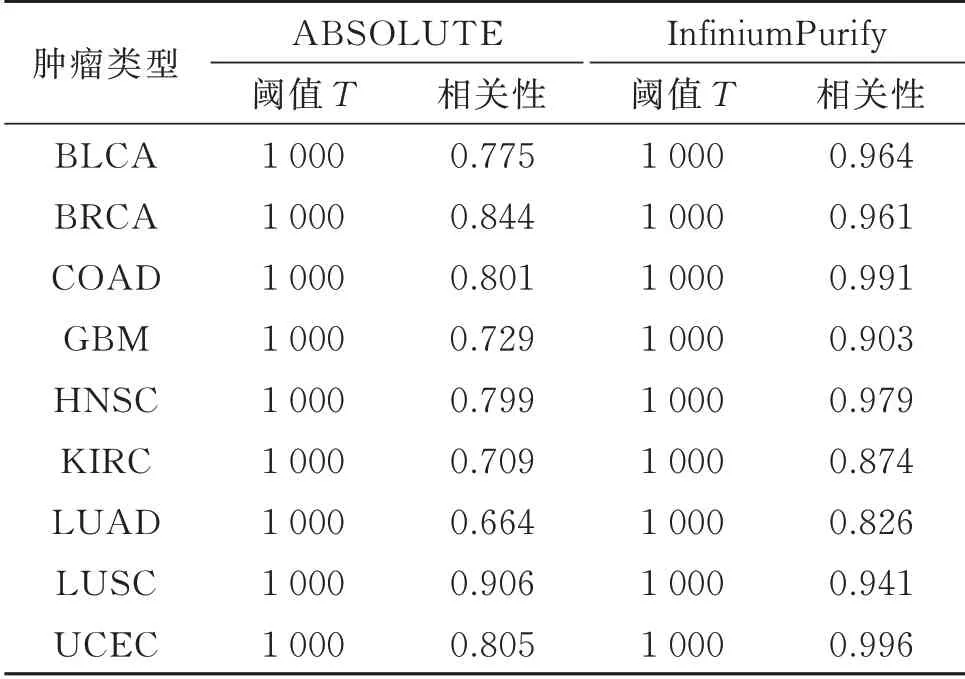
表2 T=1 000 时的相关系数表Table 2 Table of correlations with T=1 000
